An improved version of this tool is available here: https://github.com/bergmanlab/ngs_te_mapper2
ngs_te_mapper is an R implementation of the method for detecting transposable element (TE) insertions from next-generation sequencing (NGS) data published in Linheiro and Bergman (2012) PLoS ONE 7(2): e30008. The original method only detected non-reference (aka de novo) TE insertions, however it has been extended to identify TE insertions also found in the reference genome. Additionally, the current implementation uses BWA as a short read mapping engine instead of BLAT, as was used originally in Linheiro and Bergman (2012).
Non-reference TE insertions are detected using a two-stage process that relies on the presence of target site duplications (TSDs) in the region flanking the TE insertion. An overview of the two-stage mapping procedure is shown below, and is taken from Figure 1 of Linheiro and Bergman (2012).
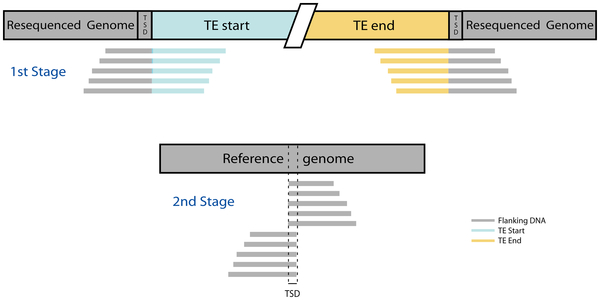
In the first stage, raw reads from a whole genome shotgun sequence are used to query against a library of reference TE sequences. 'Junction reads' that span the start/end of TE and genomic flanking sequences are retained. Such reads are often referred as 'split-reads', although in reality these reads are not split in the resequenced genome.
In the second stage, unmodified junction reads identified in the first stage are given new IDs containing information about their alignment to the TE library and then aligned to the reference genome. Alignments of the genomic positions of the unique (i.e. non-TE) components of junction reads are clustered according to whether the unique regions are 5' or 3' to a putative TE insertion site in the genome. The region of overlap between 5' and 3' clusters of junction reads defines the location of the target site duplication (TSD) for non-reference TE insertions. The orientation of the TE is then determined from the relative orientation of alignments of the junction reads to the reference genome and TE library.
Non-reference TE insertion sites are annotated as the span of TSD on zero-based, half-open coordinates and orientation is assigned in the strand field, following the framework described in Bergman (2012) Mob Genet Elements. 2:51-54.
Reference TE insertions are detected using a similar strategy to non-reference insertions, independently of any reference TE annotation. The first stage in detecting reference TE insertions is identical to the first stage of detecting non-reference TE insertions described above. The second stage in identifying reference TE insertions involves alignment of the renamed, but otherwise unmodified, junction reads to the reference genome. Alignments of the complete junction read (i.e. non-TE and TE components) are clustered to identify the two ends of the reference TE insertion. The orientation of the reference TE is then determined from the relative orientation of alignments of the junction reads to the reference genome and TE library.
In addition to the TE insertion mapping code, we provide an R script (ngs_te_logo.R) that combines TSD information from non-referenece insertions of the same TE family and outputs a sequence logo describing the local nucleotide preferences in a window around the TSD, as in Figure 3 of Linheiro and Bergman (2012). As described in the paper, the TSD logo method only works for TE families that generate a fixed length TSD (e.g. LTR retrotransposons and TIR transposons).
We note that the current version of ngs_te_logo.R does not currently filter for the modal TSD length, and thus gives slightly different results to those reported in Linheiro and Bergman (2012).
To run the mapping and logo methods on an test dataset, clone this repository and execute the test script as follows:
git clone https://github.com/bergmanlab/ngs_te_mapper.git
cd ngs_te_mapper
bash sourceCode/run_ngs_te_mapper.sh
This test script runs the main script ngs_te_mapper.R which takes six required arguments as input:
- sample (full path to fasta or fastq files of short read sequences; alternatively list of file names if the input folder option is specified, see below)
- genome (full path to reference genome fasta file)
- teFile (full path TE fasta file)
- tsd (maximum size of potential target site duplication, default=20)
- thread (number of thread used for bwa, default=1)
- output (full path to output folder)
- sourceCodeFolder (full path to location of
ngs_te_mapper.Rscript)
This test script then runs the logo script ngs_te_logo.R which takes six required arguments as input:
- genome (full path to reference genome fasta file)
- output (directory to output pdf file)
- inputFolder (location of .bed files from ngs_te_mapper.R)
- outputFile (.bed file with concatenated results from .bed files of individual samples in inputFolder)
- window (number of nucleotides analyzed upstream and downstream of TSD, default=25)
- sourceCodeFolder (full path to location of
ngs_te_mapper.Rscript)
ngs_te_mapper creates a main output directory called 'analysis'. Inside this directory there will be five other directories:
- aligned_te (.sam files of short reads aligned to the TE file, one per input sample)
- selected_reads (.fasta file of the selected reads, one file containing selected reads from all samples)
- aligned_genome (.sam file of selected reads aligned to reference genome, one file containing selected reads from all samples)
- bed_tsd (.bed file, one file containing predicted reference and non-reference TE insertions)
- logo (.pdf file, one file with one per logo per family derived from non-reference insertions)