Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomic data.
Single-cell technologies enable high-resolution studies of phenotype-defining molecular mechanisms. However, data sparsity and cellular heterogeneity make modeling biological variability across single-cell samples difficult. We present SCORPION, a tool that uses a message-passing algorithm to reconstruct comparable gene regulatory networks from single cell/nuclei RNA-seq data that are suitable for population-level comparisons (statistical analyses to compare groups consisting of multiple samples) by leveraging the same baseline priors. Using synthetic data, we found that SCORPION outperforms 12 other gene regulatory network reconstruction techniques. Using supervised experiments, we show that SCORPION can accurately identify differences in regulatory networks between wild-type and transcription factor-perturbed cells. We demonstrate SCORPION's scalability to population-level analyses using a single-cell RNA-seq atlas containing 200,436 cells from colorectal cancer and adjacent healthy tissues. The differences between tumor regions detected by SCORPION are consistent across multiple cohorts as well as with our understanding of disease progression, and elucidate phenotypic regulators that may impact patient survival.
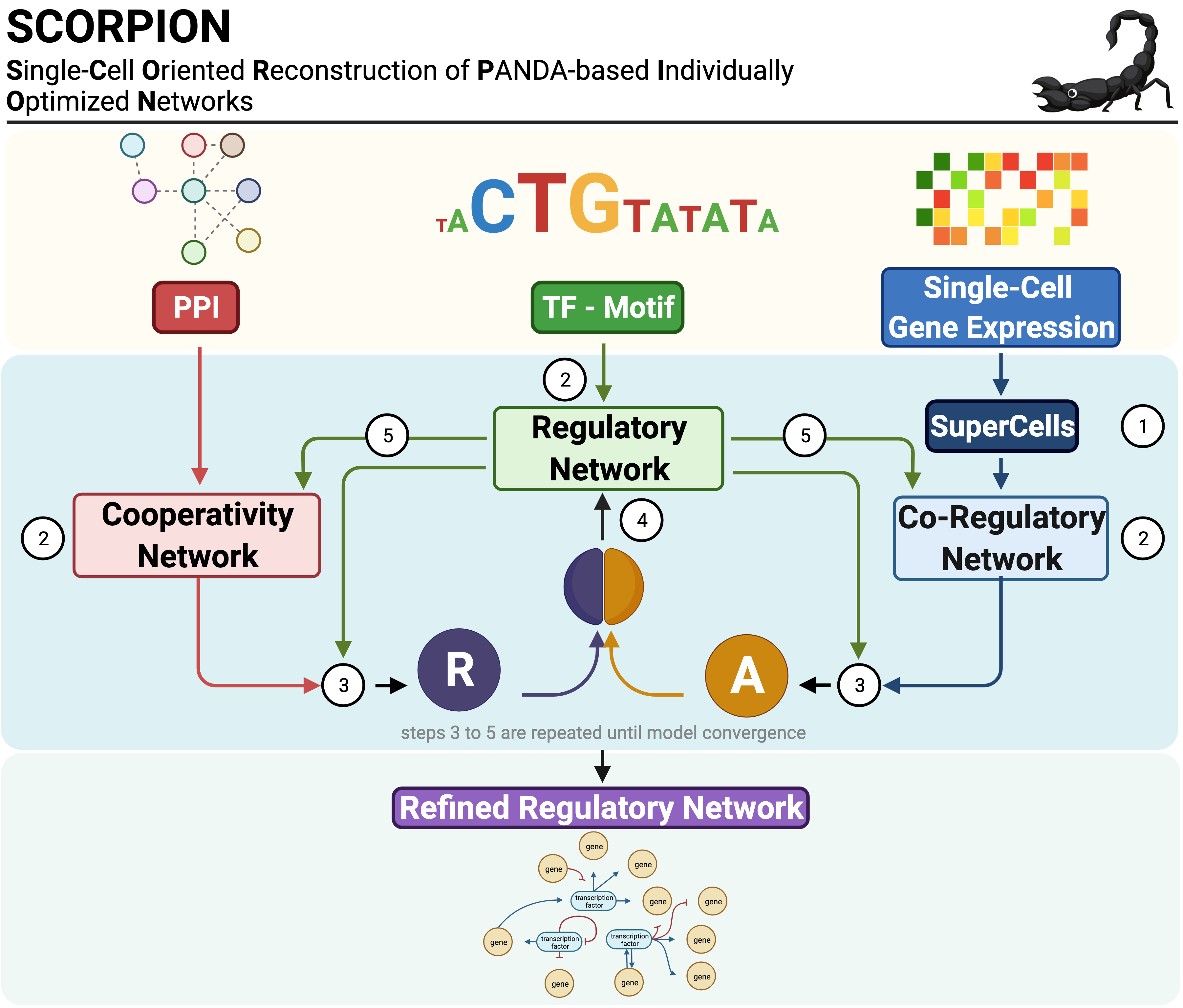
SCORPION (Single-Cell Oriented Reconstruction of PANDA Individually Optimized Gene Regulatory Networks), is an R package that uses coarse-graining of single-cell/nuclei RNA-seq data to reduce sparsity and improve the ability to detect the gene regulatory network's underlying correlation structure. The coarse-grained data generated is then used to reconstruct the gene regulatory network using a network refinement strategy through the PANDA (Passing Attributes between Networks for Data Assimilation) message passing algorithm. This algorithm is designed to integrate multiple sources of information such as protein-protein interaction, gene expression, and sequence motif data to predict accurate regulatory relationships. Thanks to the use of the same baseline priors in each instance, this approach can reconstruct comparable, fully-connected, weighted, and directed transcriptome-wide single-cell gene regulatory networks suitable for use in population-level studies.
SCORPION is available through the CRAN repositories, you can install and load it, using the following command:
install.packages('SCORPION')
library(SCORPION)
We provide an example dataset (formally a list) containing three objects. The motif data.frame describes a set of pairwise connections where a specific known sequence motif of a transcription factor was found upstream of the corresponding gene. For this particular example, the data is a subset of the transcription-factor and target gene pairs provided by the dorothea package for Homo sapiens. The expression dgCMatrix is a set of 230 gene expression levels measured across 80 PBMC cells provided by the Seurat package as pbmc_small. Finally, the ppi data.frame describes a set of known pairwise protein-protein interactions.
data(scorpionTest)
The structure of the data can be accessed as follows:
str(scorpionTest)
# List of 3
# $ gex:Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
# .. ..@ i : int [1:4456] 1 5 8 11 22 30 33 34 36 38 ...
# .. ..@ p : int [1:81] 0 47 99 149 205 258 306 342 387 423 ...
# .. ..@ Dim : int [1:2] 230 80
# .. ..@ Dimnames:List of 2
# .. .. ..$ : chr [1:230] "MS4A1" "CD79B" "CD79A" "HLA-DRA" ...
# .. .. ..$ : chr [1:80] "ATGCCAGAACGACT" "CATGGCCTGTGCAT" "GAACCTGATGAACC" "TGACTGGATTCTCA" ...
# .. ..@ x : num [1:4456] 1 1 3 1 1 4 1 5 1 1 ...
# .. ..@ factors : list()
# $ tf :'data.frame': 4485 obs. of 3 variables:
# ..$ tf : chr [1:4485] "ADNP" "ADNP" "ADNP" "AEBP2" ...
# ..$ target: chr [1:4485] "PRF1" "TMEM40" "TNFRSF1B" "CFP" ...
# ..$ mor : num [1:4485] 1 1 1 1 1 1 1 1 1 1 ...
# $ ppi:'data.frame': 12754 obs. of 3 variables:
# ..$ X.node1 : chr [1:12754] "ADNP" "ADNP" "ADNP" "AEBP2" ...
# ..$ node2 : chr [1:12754] "ZBTB14" "NFIA" "CDC5L" "YY1" ...
# ..$ combined_score: num [1:12754] 0.769 0.64 0.581 0.597 0.54 0.753 0.659 0.548 0.59 0.654 ...
Here, we are running SCORPION with large alphaValue = 0.8 for testing purposes. The default value of the alphaValue is 0.1.
scorpionOutput <- scorpion(tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
alphaValue = 0.8)
# ── SCORPION ────────────────────────────────────────────────────────────────
# ✔ Initializing and validating
# ✔ Verified sufficient samples
# ℹ Normalizing networks
# ℹ Learning Network
# ℹ Using tanimoto similarity
# ✔ Successfully ran SCORPION on 214 Genes and 783 TFs
# ℹ Time elapsed: 2.72 seconds
The structure of the output can be accessed as follows:
str(scorpionOutput)
# List of 6
# $ regNet :Formal class 'dgeMatrix' [package "Matrix"] with 4 slots
# .. ..@ x : num [1:167562] -0.413 1.517 -1.311 0.364 -1.041 ...
# .. ..@ Dim : int [1:2] 783 214
# .. ..@ Dimnames:List of 2
# .. .. ..$ : chr [1:783] "ADNP" "AEBP2" "AIRE" "ALX1" ...
# .. .. ..$ : chr [1:214] "ACAP1" "ACRBP" "ACSM3" "ADAR" ...
# .. ..@ factors : list()
# $ coregNet:Formal class 'dgeMatrix' [package "Matrix"] with 4 slots
# .. ..@ x : num [1:45796] 7.07e+06 -4.06 1.76e+01 -1.16e+01 -1.62e+01 ...
# .. ..@ Dim : int [1:2] 214 214
# .. ..@ Dimnames:List of 2
# .. .. ..$ : chr [1:214] "ACAP1" "ACRBP" "ACSM3" "ADAR" ...
# .. .. ..$ : chr [1:214] "ACAP1" "ACRBP" "ACSM3" "ADAR" ...
# .. ..@ factors : list()
# $ coopNet :Formal class 'dgeMatrix' [package "Matrix"] with 4 slots
# .. ..@ x : num [1:613089] 5.65e+06 -5.16 -3.79 -3.63 2.94 ...
# .. ..@ Dim : int [1:2] 783 783
# .. ..@ Dimnames:List of 2
# .. .. ..$ : chr [1:783] "ADNP" "AEBP2" "AIRE" "ALX1" ...
# .. .. ..$ : chr [1:783] "ADNP" "AEBP2" "AIRE" "ALX1" ...
# .. ..@ factors : list()
# $ numGenes: int 214
# $ numTFs : int 783
# $ numEdges: int 167562
This repository houses code for reproducing the analysis and figures from Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomic data. In the Code folder, files are organized for each figure —main figures (F*.R) and supplementary figures (SF*.R). To regenerate the single-cell RNA-seq colorectal atlas and the gene regulatory networks for each sample and cell-type, use the code in Atlas.R. The Data folder contains priors for analysis (human: hg38_*.RData, mouse: mm10_*.RData) and side information for atlas samples (sideList.csv). The Results folder comprises all numeric results and figures from the manuscript. The Results folder includes all numeric results and figures from the manuscript, with supplementary tables available in Supplementary Material. For the SCORPION source code, visit the Kuijjer Lab GitHub Repository. Multiplatform binaries can be found on CRAN Repositories