The AWS infrastructure for hosting a private instance (see link below) of Nextflow Tower and executing Nextflow workflows is defined in this repository and deployed using CloudFormation via Sceptre.
Click the link below and login with your @sagebase.org Google account:
➡️ Nextflow Tower @ Sage Bionetworks ⬅️
Follow the Tower User Onboarding instructions below. Access is currently restricted to Sage Bionetworks staff. See below for how to get help if you run into any issues.
Read through the contribution guidelines for more information. Contributions are welcome from anyone!
Message us in the #workflow_users Slack channel or email us at nextflow-admins[at]sagebase[dot]org.
Before you can use Nextflow Tower, you need to first deploy a Tower project, which consists an encrypted S3 bucket and the IAM resources (i.e. users, roles, and policies) that Tower requires to access the encrypted bucket and execute the workflow on AWS Batch. Once these resources exist, they need to be configured in Nextflow Tower, which is a process that has been automated using CI/CD.
-
Determine what is known as the stack name by concatenating the project name with the suffix
-project(e.g.imcore-project,amp-ad-project,commonmind-project).N.B.: Anytime that
<stack_name>appears below with the angle brackets, replace the placeholder with the actual stack name, omitting any angle brackets. -
Create an IT JIRA ticket requesting membership to the following JumpCloud groups for anyone who needs read/write or read-only access to the S3 bucket:
aws-sandbox-developersaws-workflow-nextflow-tower-viewer
To confirm whether you're already a member of these JumpCloud groups, you can expand the AWS Account list on this page (after logging in with JumpCloud) and check if you have
Developerlisted underorg-sagebase-sandboxandTowerViewerunderworkflows-nextflow-devandworkflows-nextflow-prod.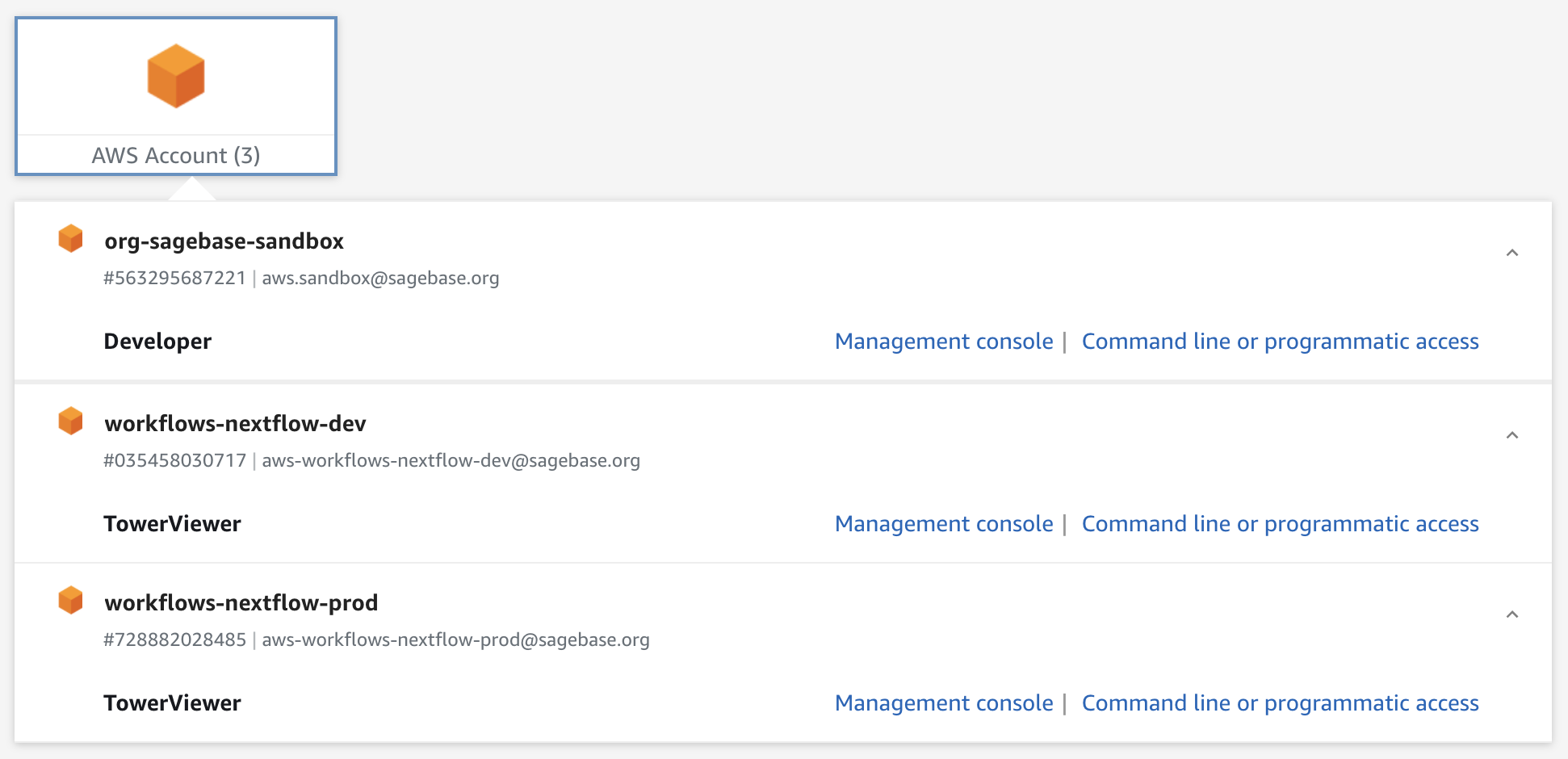
-
Open a pull request on this repository in which you duplicate
config/projects/example-project.yamlas<stack_name>.yamlin theprojects/subdirectory and then follow the numbered steps listed in the file. Note that some steps are required whereas others are optional.N.B. Here, read/write vs read-only access refers to the level of access granted to users for the encrypted S3 bucket and to the Tower workspace (more details below). Given that access is granted to the entire bucket, you might want to create more specific Tower projects that provide more granular access control.
Getting Help: If you are unfamiliar with Git/GitHub or don't know how to open a pull request, see above for how to get help.
-
Once the pull request is approved and merged, confirm that your PR was deployed successfully. If so, the following happened on your behalf:
-
All users listed under
S3ReadWriteAccessArnsandS3ReadOnlyAccessArnswere added to the Sage Bionetworks organization in Tower. -
A new Tower workspace called
<stack_name>was created under this organization. -
Users listed under
S3ReadWriteAccessArnswere added as workspace participants with theMaintainrole, which grants the following permissions:The users can launch pipeline and modify pipeline executions (e.g. can change the pipeline launch compute env, parameters, pre/post-run scripts, nextflow config) and create new pipeline configuration in the Launchpad. The users cannot modify Compute env settings and Credentials
-
Users listed under
S3ReadOnlyAccessArnswere added as workspace participants with theViewrole, which grants the following permissions:The users can access to the team resources in read-only mode
-
A set of AWS credentials called
<stack_name>was added under this Tower workspace. -
An AWS Batch compute environment called
<stack_name> (default)was created using these credentials with a default configuration that should satisfy most use cases.
N.B. If you need have special needs (e.g. more CPUs, on-demand EC2 instances, FSx for Lustre), see above for how to contact the administrators, who can create additional compute environments in your workspace.
-
-
Log into Nextflow Tower using the link at the top of this README and open your project workspace. If you were listed under
S3ReadWriteAccessArns, then you'll be able to add pipelines to your workspace and launch them on your data.

This repository is licensed under the Apache License 2.0.
Copyright 2021 Sage Bionetworks