Nextflow pipeline (DSL2) that runs fstatistics with AdmixTools, taking different input types (VCF, PLINK, EIGENSTRAT).

- Consider this tree to set popA - popD for qpDstat:
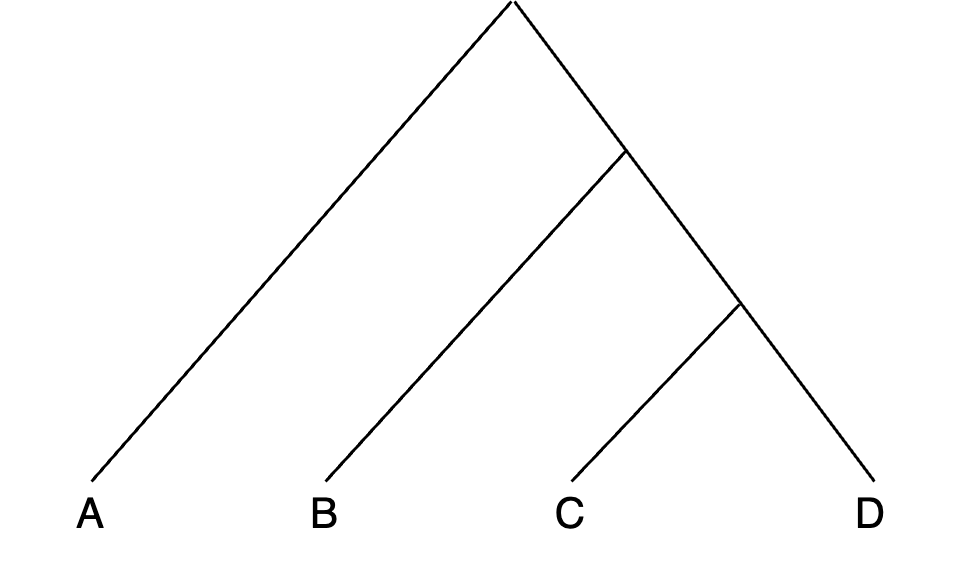
- Consider this tree to set popA - popC for qp3Pop:
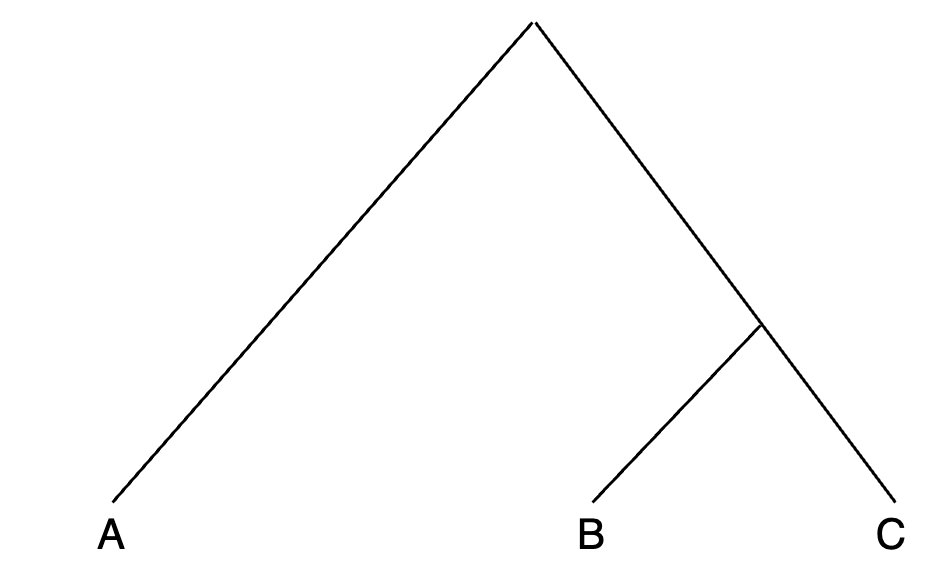


-v 0.1.0
- Supports compressed VCF file as input.
- Supports PLINK files as input.
- Supports EIGENSTRAT files as input.
- Option to run qpDstat for D or f4.
- Option to run qp3Pop for inbreed or outgroup.
- Makes all possible combination of treest for testing.
- Scalability and reproducibility via a Nextflow-based framework.
- Ubuntu 20.04.4 LTS
- macOS
| Requirement | Version | Required Commands * |
|---|---|---|
| bcftools | 1.17 | reheader,view |
| Nextflow | 23.04.2.5870 | nextflow |
| plink | 1.19 ) | plink |
| Poseidon | v1.4.0.2 | trident |
| AdmixTools | v651 and v980 | qp3Pop and qpDstat |
* These commands must be accessible from your $PATH (i.e. you should be able to invoke them from your command line).
Download nf-haplotype-selection from Github repository:
git clone https://github.com/jbv2/run_admixtools.git
To test run_admixtools execution using test data, run:
nextflow run main.nf -profile <test_qpDstat,test_qp3Pop>This pipeline can use nf-core Institutional profiles. For DAG EVA people, please use:
nextflow run main.nf -profile eva,archgen,<test_qpDstat,test_qp3Pop>To run run_admixtools go to the pipeline directory and execute:
nextflow run main.nf \
--inputVCF "<path to VCF input>" \ # Needs --input_type "vcf" --samples and --half_call
--samples <Path to file with new sample names> \ # txt file with id famid_id needed when input is VCF.
--half_call "<h>" \ # Plink option when VCF to PLINK on how to treat how to deal with '0/.'. See https://www.cog-genomics.org/plink/1.9/input
--inputbed "<path to PLINK bed>" \ # Needs input_type = "plink"
--inputgeno "<Path to EIGENSTRAT geno>" \ # Needs input_type = "eigenstrat"
--input_type <'vcf','plink','eigenstrat'> \ # Select your input type
--popA "<Vector for population(s) in A position>" \ # Vector. Example: "CEU YRI"
--popB "<Vector for population(s) in B position>" \ # Vector. Example: "CEU YRI"
--popC "<Vector for population(s) in C position>" \ # Vector. Example: "CEU YRI"
--popD "<Vector for population(s) in D position>" \ # Vector. Example: "CEU YRI". Not needed in qp3Pop.
--run_qpDstat <'true','false'> \ # Can not be used with --run_qp3Pop. Needs --f4mode
--f4mode <"YES","NO"> \ # qpDstat option
--run_qp3Pop <'true','false'> \ # Can not be used with --run_qpDstat. Needs --inbreed.
--inbreed <"YES","NO"> \ # qp3Pop option
--outdir <path to results> \ # Outdir -
If you have one input type, you don't need to provide the others.
-
If you select PLINK as input,
.famand.bimmust be in the same dir as the.bedfile. -
If you select EIGENSTRAT as input,
.indand.snpmust be in the same dir as the.genofile. -
If you select VCF as input, provide:
- --samples "Two columns text file with the structure: ID FAMID_ID". See
test/reference/new_sample_names.txtfor deatils. - --half_call: Plink option when VCF to PLINK on how to treat how to deal with '0/.'. Default: haploid.
- 'haploid'/'h': Treat half-calls as haploid/homozygous (the PLINK 1 file format does not distinguish between the two). This maximizes similarity between the VCF and BCF2 parsers.
- 'missing'/'m': Treat half-calls as missing.
- 'reference'/'r': Treat the missing part as reference.
- --samples "Two columns text file with the structure: ID FAMID_ID". See
You can also copy/rename test/my_project.config, replace with your information and run it with:
nextflow run main.nf -c </PATH/TO/YOUR/my_projecy.config> -profile my_project_f4 It can be as easy as you want!
- If you have a VCF to start:
- A
VCFfile compressed with a.vcf.gzextension. AVCFfile mainly contains meta-information lines, a header and data lines with information about each position. The header names the eigth mandatory columnsCHROM, POS, ID, REF, ALT, QUAL, FILTER, INFO.
For more information about the VCF format, please go to the next link: Variant Call Format
Example line(s):
##fileformat=VCFv4.2
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1 sample2 sample3
21 9411773 rs867796868 C T . PASS AC=0;AN=182 GT 0|0 0|0 0|0 0|0
21 10979896 rs117219976 A G . PASS AC=1;AN=182 GT 0|1 0|0 0|0
22 17176028 rs2845365 T G . PASS AC=58;AN=182 GT 1|0 0|0 0|1 - A two columns file, separated by space with old_name new_name.
sample1 FAM1_sample1
sample2 FAM2_sample2
sample3 FAM3_sample3- If you have bfile (PLINK) to start:
- A binary
.bed. - A
.bimfile.
Example line(s):
1 rs74512038 0 778597 T C
1 rs553642122 0 790021 T C
1 rs4951859 0 794299 G C
...- A
.famfile.
Example line(s):
FAM1 sample1 0 0 0 -9
FAM2 sample2 0 0 0 -9
FAM3 sample3 0 0 0 -9
...- If you have EIGENSTRAT to start:
- A binary
.geno. - A
.snpfile.
Example line(s):
rs74512038 1 0 778597 T C
rs553642122 1 0 790021 T C
rs4951859 1 0 794299 G C
...- A
.indfile.
Example line(s):
sample1 U FAM1
sample2 U FAM2
sample3 U FAM3
...For qp3Pop
- A
.OUT.logfile.
Example line(s):
qp3Pop: parameter file: test.parfile
### THE INPUT PARAMETERS
##PARAMETER NAME: VALUE
genotypename: test.geno
snpname: test.snp
indivname: test.ind
popfilename: test.ftest.txt
inbreed: YES
## qp3Pop version: 651
inbreed set YES
test.snp: genetic distance set from physical distance
*** warning: first snp . is number. perhaps you are using .map format
number of triples 3
nplist: 3
snps: 59920
number of blocks for block jackknife: 9
Source 1 Source 2 Target f_3 std. err Z SNPs
result: CHB PEL Maya 0.001641 0.011348 0.145 53604- A
.ftest.txtfile with combinations.
Example line(s):
CHB PEL Maya
CHB PEL Mixe
CHB PEL Nahua- A
.parfilefile.
Example line(s):
genotypename: test.geno
snpname: test.snp
indivname: test.ind
popfilename: test.ftest.txt
inbreed: YESFor qpDstat
- A
.OUT.logfile.
Example line(s):
qpDstat: parameter file: test.parfile
### THE INPUT PARAMETERS
##PARAMETER NAME: VALUE
genotypename: test.geno
snpname: test.snp
indivname: test.ind
popfilename: test.ftest.txt
printsd: YES
f4mode: YES
## qpDstat version: 980
inbreed set NO
test.snp: genetic distance set from physical distance
*** warning: first snp . is number. perhaps you are using .map format
number of quadruples 6
0 CHB 1
1 PEL 4
2 Maya 4
3 Mixe 5
4 Nahua 13
jackknife block size: 0.050
snps: 59920 indivs: 27
number of blocks for block jackknife: 9
nrows, ncols: 27 59920
result: CHB PEL Maya Mixe -0.000913 0.003262 -0.280 3756 3811 59905
result: CHB PEL Maya Nahua 0.002345 0.002936 0.799 3875 3734 59905
result: CHB PEL Mixe Maya 0.000913 0.003262 0.280 3811 3756 59905 - A
.result.tsvwith header and separated by TAB.
PopA PopB PopC PopD f4 stderr Zscore
CHB PEL Maya Mixe -0.000913 0.003262 -0.280
CHB PEL Maya Nahua 0.002345 0.002936 0.799
CHB PEL Mixe Maya 0.000913 0.003262 0.280
CHB PEL Mixe Nahua 0.003258 0.001824 1.787
CHB PEL Nahua Maya -0.002345 0.002936 -0.799
CHB PEL Nahua Mixe -0.003258 0.001824 -1.787- A
.ftest.txtfile with combinations.
Example line(s):
CHB PEL Maya Mixe
CHB PEL Maya Nahua
CHB PEL Mixe Maya
CHB PEL Mixe Nahua
CHB PEL Nahua Maya
CHB PEL Nahua Mixe- A
.parfilefile.
Example line(s):
genotypename: test.geno
snpname: test.snp
indivname: test.ind
popfilename: test.ftest.txt
printsd: YES
f4mode: YESUnder the hood run_admixtools uses some coding tools, please include the following ciations in your work:
- Narasimhan, V., Danecek, P., Scally, A., Xue, Y., Tyler-Smith, C., & Durbin, R. (2016). BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics, 32(11), 1749-1751.
- Patterson, Nick, et al. "Ancient admixture in human history." Genetics 192.3 (2012): 1065-1093.
- Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., ... & Sham, P. C. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. The American journal of human genetics, 81(3), 559-575.
- https://github.com/poseidon-framework/poseidon-hs
If you have questions, requests, or bugs to report, please email judith.vballesteros@gmail.com
Judith Ballesteros-Villascán judith.vballesteros@gmail.com