
| Overview: | A set of tools to help building or using Sequana pipelines |
|---|---|
| Status: | Production |
| Issues: | Please fill a report on github |
| Python version: | Python 3.7, 3.8, 3.9 |
| Citation: | Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352 |
sequana_pipetools is a set of tools to help us managing the Sequana pipelines (NGS pipelines such as RNA-seq, Variant, ChIP-seq, etc).
The goal of this package is to make the deployment of Sequana pipelines easier by moving some of the common tools used by the different pipelines in a pure Python library.
The Sequana framework used to have all bioinformatics, snakemake rules, pipelines, tools to manage pipelines in a single library (Sequana) as described in Fig 1 here below.
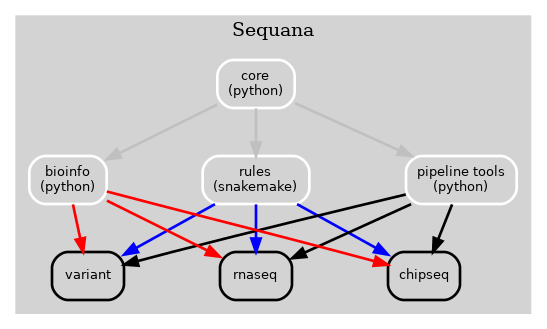
Figure 1 Old Sequana framework will all pipelines and Sequana library in the same place including pipetools (this library).
Each time we changed anything, the entire library needed to be checked carefully (even though we had 80% test coverage). Each time a pipeline was added, new dependencies woule be needed, and so on. So, we first decided to make all pipelines independent as shown in Fig 2:
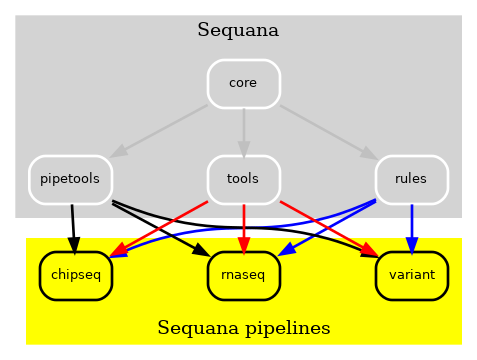
Figure 2 v0.8 of Sequana moved the Snakemake pipelines in indepdendent repositories. A cookie cutter ease the creation of scuh pipelines
That way, we could change a pipeline without the need to update Sequana, and vice-versa. This was already a great jump ahead. Yet, some tools reprensented here by the pipetools box were required by all pipelines. This was mostly for providing user interface, sanity check of input data, etc. This was moving fast with new pipelines added every month. To make the pipelines and Sequana more modular, we decided to create a pure Python library that would make the pipelines even more independent as shown in Fig3. We called it sequana_pipetools.
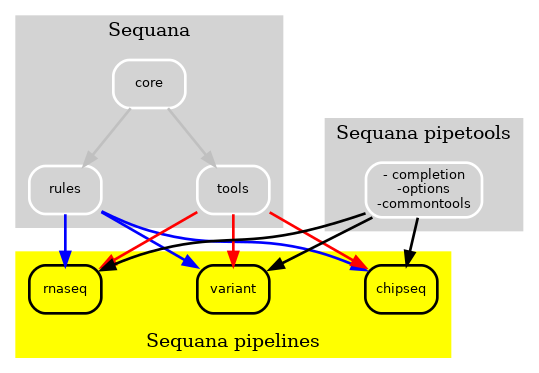
Figure 3 New Sequana framework. The library itself with the core, the bioinformatics tools is now independent of the pipelines. Besides, the pipetools library provide common tools to all pipelines to help in their creation/management. For instance, common parser for options.
from pypi website:
pip install sequana_pipetools
No dependencies for this package except Python itself. In practice, this package has no interest if not used with a Sequana pipeline. So, when using it, you will need to install the relevant Sequana pipelines that you wish to use.
This package is for Sequana developers. To get more help, go to the doc directory and build the local sphinx directory using:
make html browse build/html/index.html
There are currently two standalone tools. The first one is for Linux users under bash to obtain completion of a sequana pipeline command line arguments:
sequana_completion --name fastqc
The second is used to introspect slurm files to get a summary of the SLURM log files:
sequana_slurm_status --directory .
Will print a short summary report with common errors (if any).
The library is intended to help Sequana developers to design their pipelines. See the Sequana organization repository for examples.
In addition to those standalones, sequana_pipetools goal is to provide utilities to help Sequana developers. We currently provide a set of Options classes that should be used to design the API of your pipelines. For example, the sequana_pipetools.options.SlurmOptions can be used as follows inside a standard Python module (the last two lines is where the magic happens):
import argparse
from sequana_pipetools.options import *
from sequana_pipetools.misc import Colors
from sequana_pipetools.info import sequana_epilog, sequana_prolog
col = Colors()
NAME = "fastqc"
class Options(argparse.ArgumentParser):
def __init__(self, prog=NAME, epilog=None):
usage = col.purple(sequana_prolog.format(**{"name": NAME}))
super(Options, self).__init__(usage=usage, prog=prog, description="",
epilog=epilog,
formatter_class=argparse.ArgumentDefaultsHelpFormatter
)
# add a new group of options to the parser
so = SlurmOptions()
so.add_options(self)
Developers should look at e.g. module sequana_pipetools.options for the API reference and one of the official sequana pipeline (e.g., https://github.com/sequana/sequana_variant_calling) to get help from examples.
The Options classes provided can be used and combined to design pipelines. The code from sequana_pipetools is used within our template to automatically create pipeline tree structure using a cookie cutter. This cookie cutter is available in https://github.com/sequana/sequana_pipeline_template and as a standalone in Sequana itself (sequana_init_pipeline).
Sequana is a versatile tool that provides
- A Python library dedicated to NGS analysis (e.g., tools to visualise standard NGS formats).
- A set of Pipelines dedicated to NGS in the form of Snakefiles (Makefile-like with Python syntax based on snakemake framework) with more than 80 re-usable rules.
- Standalone applications.
See the sequana home page for details.
To join the project, please let us know on github.
| Version | Description |
|---|---|
| 0.8.0 |
|
| 0.7.6 |
|
| 0.7.5 |
|
| 0.7.4 |
|
| 0.7.3 |
|
| 0.7.2 |
|
| 0.7.1 |
|
| 0.7.0 |
|
| 0.6.3 |
|
| 0.6.2 |
|
| 0.6.1 |
|
| 0.6.0 |
|
| 0.5.3 |
|
| 0.5.2 |
|
| 0.5.1 |
|
| 0.5.0 |
|
| 0.4.3 |
|
| 0.4.2 |
|
| 0.4.1 |
|
| 0.4.0 |
|
| 0.3.1 |
|
| 0.3.0 |
|
| 0.2.6 |
|
| 0.2.5 |
|
| 0.2.4 |
|
| 0.2.3 |
|
| 0.2.2 |
|
| 0.2.1 |
|
| 0.2.0 | add content from sequana.pipeline_common to handle all kind of options in the argparse of all pipelines. This is independent of sequana to speed up the --version and --help calls |
| 0.1.2 | add version of the pipeline in the output completion file |
| 0.1.1 | release bug fix |
| 0.1.0 | creation of the package |