SomaticSeq: An ensemble approach to accurately detect somatic mutations
- Detailed documentation is included in the package. It's located in docs/Manual.pdf. Quick guide can be found here.
- SomaticSeq's open-access paper published in Genome Biology.
- Feel free to report issues and/or ask questions at the Issues page.
- Note: Do not worry if Python throws the following warning. This occurs when SciPy attempts a statistical test with empty data. This is expected when there is no variant read in the matched normal, resulting in NaN in the output.
RuntimeWarning: invalid value encountered in double_scalars z = (s - expected) / np.sqrt(n1*n2*(n1+n2+1)/12.0)
Dockerized Pipelines
- The docker repo for SomaticSeq can be found at https://hub.docker.com/r/lethalfang/somaticseq/.
- Since v2.3.0, we have run script generators for the dockerized somatic mutation callers at utilities/dockered_pipelines. The documentation for those scripts are in Section 4 of the User's Manual.
- The pipeline to generate training data out of your own sequencing data based on BAMSurgeon is located at utilities/dockered_pipelines/bamSimulator.
- The alignment pipeline to generate BAM files based on GATK's best practices is at utilities/dockered_pipelines/alignments.
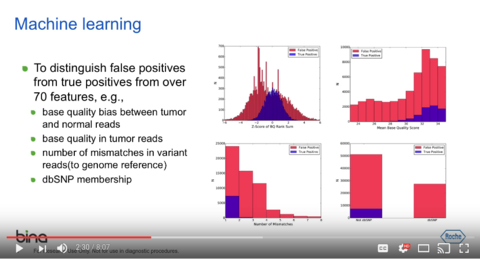
For a quick description of SomaticSeq, you may watch this 8-minute video: