



![]()
![]()
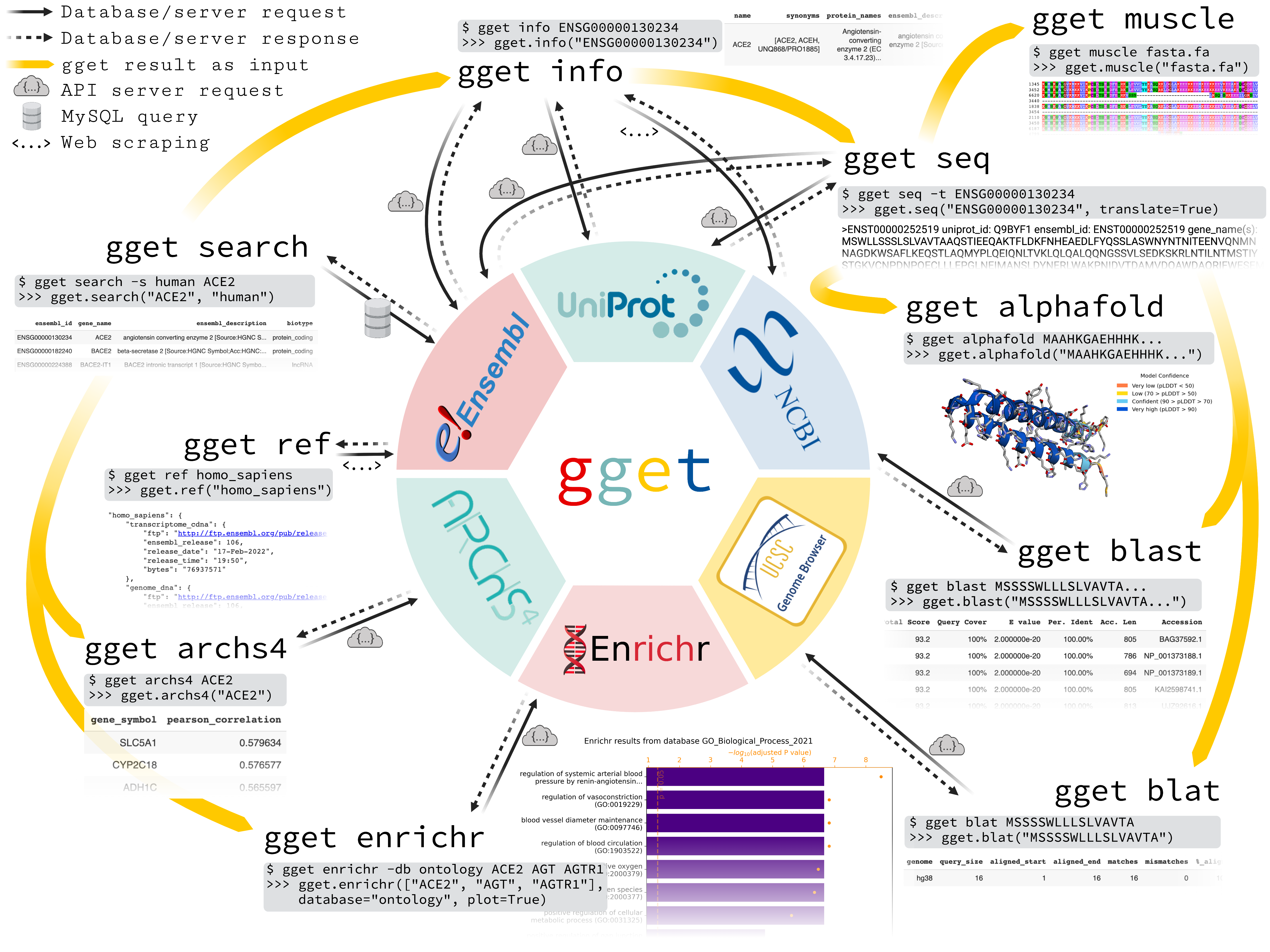
gget is a free, open-source command-line tool and Python package that enables efficient querying of genomic databases. gget consists of a collection of separate but interoperable modules, each designed to facilitate one type of database querying in a single line of code.
If you use gget in a publication, please cite*:
Luebbert, L., & Pachter, L. (2023). Efficient querying of genomic reference databases with gget. Bioinformatics. https://doi.org/10.1093/bioinformatics/btac836
Read the article here: https://doi.org/10.1093/bioinformatics/btac836
pip install --upgrade ggetAlternative:
conda install -c bioconda ggetFor use in Jupyter Lab / Google Colab:
# Python
import ggetCommand line:
# Fetch all Homo sapiens reference and annotation FTPs from the latest Ensembl release
$ gget ref homo_sapiens
# Get Ensembl IDs of human genes with "ace2" or "angiotensin converting enzyme 2" in their name/description
$ gget search -s homo_sapiens 'ace2' 'angiotensin converting enzyme 2'
# Look up gene ENSG00000130234 (ACE2) and its transcript ENST00000252519
$ gget info ENSG00000130234 ENST00000252519
# Fetch the amino acid sequence of the canonical transcript of gene ENSG00000130234
$ gget seq --translate ENSG00000130234
# Quickly find the genomic location of (the start of) that amino acid sequence
$ gget blat MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS
# BLAST (the start of) that amino acid sequence
$ gget blast MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS
# Align multiple nucleotide or amino acid sequences against each other (also accepts path to FASTA file)
$ gget muscle MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS
# Align one or more amino acid sequences against a reference (containing one or more sequences) (local BLAST) (also accepts paths to FASTA files)
$ gget diamond MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS -ref MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS
# Use Enrichr for an ontology analysis of a list of genes
$ gget enrichr -db ontology ACE2 AGT AGTR1 ACE AGTRAP AGTR2 ACE3P
# Get the human tissue expression of gene ACE2
$ gget archs4 -w tissue ACE2
# Get the protein structure (in PDB format) of ACE2 as stored in the Protein Data Bank (PDB ID returned by gget info)
$ gget pdb 1R42 -o 1R42.pdb
# Find Eukaryotic Linear Motifs (ELMs) in a protein sequence
$ gget setup elm # setup only needs to be run once
$ gget elm -o results MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS
# Fetch a scRNAseq count matrix (AnnData format) based on specified gene(s), tissue(s), and cell type(s) (default species: human)
$ gget setup cellxgene # setup only needs to be run once
$ gget cellxgene --gene ACE2 SLC5A1 --tissue lung --cell_type 'mucus secreting cell' -o example_adata.h5ad
# Predict the protein structure of GFP from its amino acid sequence
$ gget setup alphafold # setup only needs to be run once
$ gget alphafold MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYKPython (Jupyter Lab / Google Colab):
import gget
gget.ref("homo_sapiens")
gget.search(["ace2", "angiotensin converting enzyme 2"], "homo_sapiens")
gget.info(["ENSG00000130234", "ENST00000252519"])
gget.seq("ENSG00000130234", translate=True)
gget.blat("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget.blast("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget.muscle(["MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS", "MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS"])
gget.diamond("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS", reference="MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS")
gget.enrichr(["ACE2", "AGT", "AGTR1", "ACE", "AGTRAP", "AGTR2", "ACE3P"], database="ontology", plot=True)
gget.archs4("ACE2", which="tissue")
gget.pdb("1R42", save=True)
gget.setup("elm") # setup only needs to be run once
ortho_df, regex_df = gget.elm("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget.setup("cellxgene") # setup only needs to be run once
gget.cellxgene(gene = ["ACE2", "SLC5A1"], tissue = "lung", cell_type = "mucus secreting cell")
gget.setup("alphafold") # setup only needs to be run once
gget.alphafold("MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK")Call gget from R using reticulate:
system("pip install gget")
install.packages("reticulate")
library(reticulate)
gget <- import("gget")
gget$ref("homo_sapiens")
gget$search(list("ace2", "angiotensin converting enzyme 2"), "homo_sapiens")
gget$info(list("ENSG00000130234", "ENST00000252519"))
gget$seq("ENSG00000130234", translate=TRUE)
gget$blat("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget$blast("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget$muscle(list("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS", "MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS"), out="out.afa")
gget$diamond("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS", reference="MSSSSWLLLSLVEVTAAQSTIEQQAKTFLDKFHEAEDLFYQSLLAS")
gget$enrichr(list("ACE2", "AGT", "AGTR1", "ACE", "AGTRAP", "AGTR2", "ACE3P"), database="ontology")
gget$archs4("ACE2", which="tissue")
gget$pdb("1R42", save=TRUE)