


variation graph data structures, interchange formats, alignment, genotyping, and variant calling methods
![]()
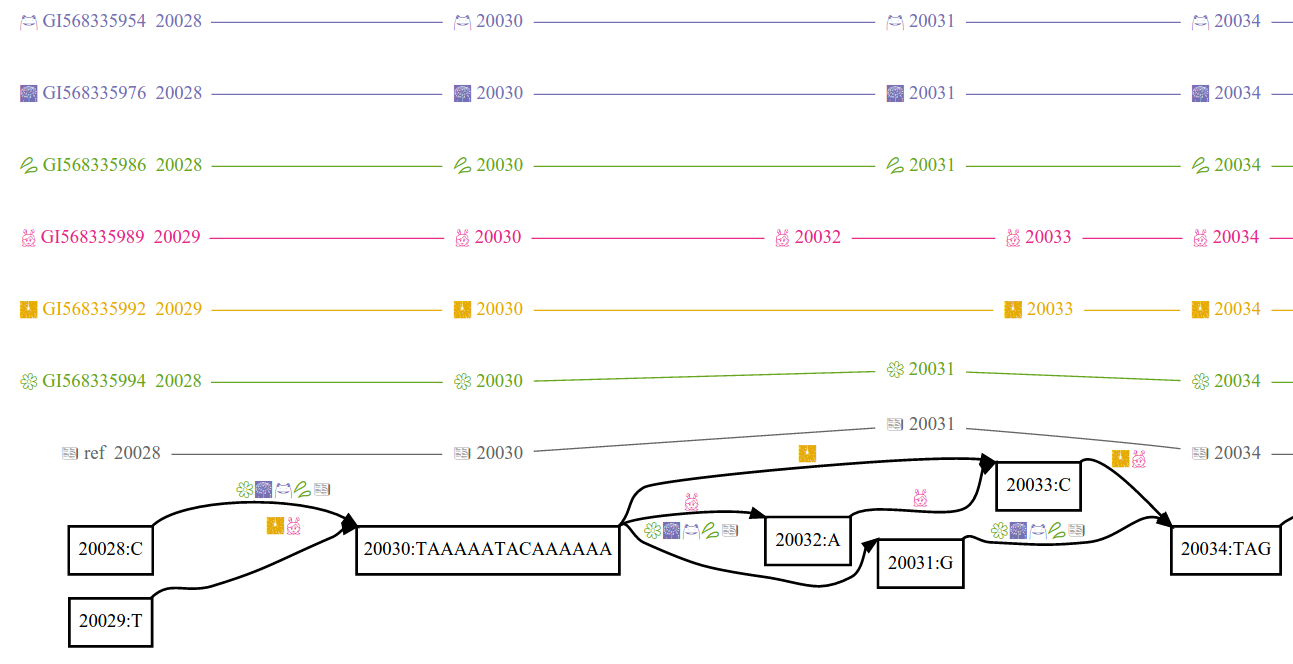
Variation graphs provide a succinct encoding of the sequences of many genomes. A variation graph (in particular as implemented in vg) is composed of:
- nodes, which are labeled by sequences and ids
- edges, which connect two nodes via either of their respective ends
- paths, describe genomes, sequence alignments, and annotations (such as gene models and transcripts) as walks through nodes connected by edges
This model is similar to sequence graphs that have been used in assembly and multiple sequence alignment.
Paths provide coordinate systems relative to genomes encoded in the graph, allowing stable mappings to be produced even if the structure of the graph is changed. The variation graph model makes this embedding explicit and essential. Tools in vg maintain paths as immutable during transformations of the graph. They use paths to project graph-relative data into reference-relative coordinate spaces. Paths provide stable coordinates for graphs built in different ways from the same input sequences.
We maintain a support forum on biostars: https://www.biostars.org/t/vg/
The easiest way to get vg is to download one of our release builds for Linux. We have a 6-week release cadence, so our builds are never too far out of date.

Download the latest vg release for Linux
For MacOS, see Building on MacOS.
If you don't want to or can't use a pre-built release of vg, or if you want to become a vg developer, you can build it from source instead.
First, obtain the repo and its submodules:
git clone --recursive https://github.com/vgteam/vg.git
cd vg
Then, install VG's dependencies. You'll need the protobuf and jansson development libraries installed, and to run the tests you will need jq, bc and rs. On Ubuntu, you should be able to do:
make get-deps
On other distros, you will need to perform the equivalent of:
sudo apt-get install build-essential git cmake pkg-config libncurses-dev libbz2-dev \
protobuf-compiler libprotoc-dev libprotobuf-dev libjansson-dev \
automake libtool jq bc rs curl unzip redland-utils \
librdf-dev bison flex gawk lzma-dev liblzma-dev liblz4-dev \
libffi-dev libcairo-dev
Note that Ubuntu 16.04 does not ship a sufficiently new Protobuf; vg requires Protobuf 3 which will have to be manually installed.
At present, you will need GCC version 4.9 or greater, with support for C++14, to compile vg. (Check your version with gcc --version.)
Other libraries may be required. Please report any build difficulties.
Note that a 64-bit OS is required. Ubuntu 18.04 should work. You will also need a CPU that supports SSE 4.2 to run VG; you can check this with cat /proc/cpuinfo | grep sse4_2.
When you are ready, build with . ./source_me.sh && make, and run with ./bin/vg.
You can also produce a static binary with make static, assuming you have static versions of all the dependencies installed on your system.
The first step is to clone the vg repository:
git clone --recursive https://github.com/vgteam/vg.git
cd vg
VG depends on a number of packages being installed on the system where it is being built. Dependencies can be installed using either MacPorts or Homebrew.
You can use MacPorts to install VG's dependencies:
sudo port install libtool protobuf3-cpp jansson jq cmake pkgconfig autoconf automake libtool coreutils samtools redland bison gperftools md5sha1sum rasqal gmake autogen cairo libomp
Homebrew provides another package management solution for OSX, and may be preferable to some users over MacPorts. VG ships a Brewfile describing its Homebrew dependencies, so from the root vg directory, you can install dependencies, and expose them to vg, like this:
# Install all the dependencies in the Brewfile
brew bundle
# Use GNU versions of coreutils over Apple versions
export PATH="/usr/local/opt/coreutils/libexec/gnubin:/usr/local/bin:$PATH"
# Force use of new version of bison
brew link bison --force
# NOTE! If brew says that it is refusing to link Bison, follow its suggested
# instructions to put Bison on your PATH instead.
# Use glibtool/ize
export LIBTOOL=glibtool
export LIBTOOLIZE=glibtoolize
# Use installed libraries
export LD_LIBRARY_PATH=/usr/local/lib:$LD_LIBRARY_PATH;
export LIBRARY_PATH=$LD_LIBRARY_PATH;
While Apple's clang can build VG, the C++ standard library it uses doesn't support some parallel extensions, so a Clang-built VG will be slower. Better results can be achieved by building with GNU GCC >= 4.9 and its libstdc++ standard library.
With MacPorts, you can install GNU GCC like this:
sudo port install gcc9 clang-8.0
To make GCC 9 the default compiler, run (use none instead of mp-gcc7 to revert back):
sudo port select gcc mp-gcc9
Some OSX users also need to have the MacPorts Clang assembler for building VG's dependencies (use none instead of mp-clang-8.0 to revert back):
sudo port select clang mp-clang-8.0
With Homebrew, you can install GNU GCC for VG like this:
brew install gcc6
# Manually create symlinks to make Homebrew GCC 6 the default gcc and g++
ln -s gcc-6 /usr/local/bin/gcc
ln -s g++-6 /usr/local/bin/g++
With dependencies and compilers installed, VG can now be built:
. ./source_me.sh && make
Note that static binaries cannot yet be built for Mac.
Our team has successfully built vg on Mac with GCC versions 4.9, 5.3, 6, 7, and 7.3, as well as Clang 9.0.
The simplest thing to do with vg is to build a graph and align to it. At present, you'll want to use a reference and VCF file to do so. If you're working in the test/ directory:
vg construct -r small/x.fa -v small/x.vcf.gz >x.vgNote that to build a graph, an index of the VCF file is required. The VCF index file can be generated using the tabix command provided by SAMtools (e.g. tabix -p vcf x.vcf.gz on the command line).
vg view provides a way to convert the graph into various formats:
# GFA output
vg view x.vg >x.gfa
# dot output suitable for graphviz
vg view -d x.vg >x.dot
# json version of binary alignments
vg view -a x.gam >x.jsonAs this is a small graph, you could align to it using a full-length partial order alignment:
vg align -s CTACTGACAGCAGAAGTTTGCTGTGAAGATTAAATTAGGTGATGCTTG x.vgNote that you don't have to store the graph on disk at all, you can simply pipe it into the local aligner:
vg construct -r small/x.fa -v small/x.vcf.gz | vg align -s CTACTGACAGCAGAAGTTTGCTGTGAAGATTAAATTAGGTGATGCTTG -Most commands allow the streaming of graphs into and out of vg.
If your graph is large, you want to use vg index to store the graph and vg map to align reads. vg map implements a kmer based seed and extend alignment model that is similar to that used in aligners like novoalign or MOSAIK. First an on-disk index is built with vg index which includes the graph itself and kmers of a particular size. When mapping, any kmer size shorter than that used in the index can be employed, and by default the mapper will decrease the kmer size to increase sensitivity when alignment at a particular k fails.
# construct the graph (paths below assume running from `vg/test` directory)
vg construct -r small/x.fa -v small/x.vcf.gz > x.vg
# store the graph in the xg/gcsa index pair
vg index -x x.xg -g x.gcsa -k 16 x.vg
# align a read to the indexed version of the graph
# note that the graph file is not opened, but x.vg.index is assumed
vg map -s CTACTGACAGCAGAAGTTTGCTGTGAAGATTAAATTAGGTGATGCTTG -x x.xg -g x.gcsa > read.gam
# simulate a bunch of 150bp reads from the graph, one per line
vg sim -n 1000 -l 150 -x x.xg > x.sim.txt
# now map these reads against the graph to get a GAM
vg map -T x.sim.txt -x x.xg -g x.gcsa > aln.gam
# surject the alignments back into the reference space of sequence "x", yielding a BAM file
vg surject -x x.xg -b aln.gam > aln.bam
# or alternatively, surject them to BAM in the call to map
vg sim -n 1000 -l 150 -x x.xg > x.sim.txt
vg map -T x.sim.txt -x x.xg -g x.gcsa --surject-to bam > aln.bamVariation from alignments can be embedded back into the graph. This process is called augmentation and is important for variant calling, for example (see below).
# augment the graph with all variation from the GAM except that implied by soft clips, saving to aug.vg. aug.gam contains the same reads as aln.gam but mapped to aug.vg
vg augment x.vg aln.gam -A aug.gam > aug.vg
# augment the graph with all variation from the GAM, saving each mapping as a path in the graph.
# softclips of alignment paths are preserved (`-S`).
# Note, this can be much less efficient than the above example if there are many alignments in the GAM
vg augment x.vg aln.gam -i -S > aug_with_paths.vgThe following examples show how to generate a VCF with vg using read support. They depend on output from the Mapping and Augmentation examples above. Small variants and SVs can be called using the same approach. Currently, it is more accuracte for SVs.
Call only variants that are present in the graph (use -g):
# Compute the read support from the gam (ignoring mapping and base qualitiy < 5)
vg pack -x x.xg -g aln.gam -Q 5 -o aln.pack
# Generate a VCF from the support.
vg call x.xg -k aln.pack > graph_calls.vcfBy default, vg call omits 0/0 variants and tries to normalize alleles to make the VCF more compact. Both these steps can make it difficult to compare the outputs from different samples as the VCFs will have different coordinates even though they were created using the same graph. The -a option addresses this by calling every snarl using the same coordinates and including reference calls. Outputs for different samples can be combined with bcftools merge -m all.
vg call x.xg -k aln.pack -a > snarl_genotypes.vcf
In order to also consider novel variants from the reads, use the augmented graph and gam (as created in the previous example using vg augment -A):
# Index our augmented graph
vg index aug.vg -x aug.xg
# Compute the read support from the augmented gam (with ignoring qualitiy < 5)
vg pack -x aug.xg -g aug.gam -Q 5 -o aln_aug.pack
# Generate a VCF from the support
vg call aug.xg -k aln_aug.pack > calls.vcfA similar process can by used to genotype known variants from a VCF. To do this, the graph must be constructed from the VCF with vg construct -a:
# Re-construct the same graph as before but with `-a`
vg construct -r small/x.fa -v small/x.vcf.gz -a > xa.vg
# Index the graph with `-L' to preserve alt paths in the xg
vg index xa.vg -x xa.xg -L
# Compute the support (we could also reuse aln.pack from above)
vg pack -x xa.xg -g aln.gam -o aln.pack
# Genotype the VCF (use -v)
vg call xa.xg -k aln.pack -v small/x.vcf.gz > genotypes.vcfPre-filtering the GAM before computing support can improve precision of SNP calling
# filter secondary and ambiguous read mappings out of the gam
vg filter aln.gam -r 0.90 -fu -m 1 -q 15 -D 999 -x x.xg > aln.filtered.gam
# then compute the support from aln.filtered.gam instead of aln.gam in above etc.For larger graphs, it is recommended to compute snarls separately:
vg snarls x.xg > x.snarls
# load snarls from a file instead of computing on the fly
vg call x.xg -k aln.pack -r x.snarls > calls.vcfNote: vg augment, vg pack, vg call and vg snarls can now all be run on directly on any graph format (ex .vg, .xg (except augment) or anything output by vg convert). Operating on .vg uses the most memory and is not recommended for large graphs. The output of vg pack can only be read in conjunction with the same graph used to create it, so vg pack x.vg -g aln.gam -o x.pack then vg call x.xg -k x.pack will not work.
Infer variants from from alignments implied by paths in the graph. This can be used, for example, to call SVs directly from a variation graph that was constructed from a multiple alignment of different assemblies:
# create a graph from a multiple alignment of HLA haplotypes (from vg/test directory)
vg msga -f GRCh38_alts/FASTA/HLA/V-352962.fa -t 1 -k 16 | vg mod -U 10 - | vg mod -c - > hla.vg
# index it
vg index hla.vg -x hla.xg
# generate a VCF using gi|568815592:29791752-29792749 as the reference contig. The other paths will be considered as haploid samples
vg deconstruct hla.xg -e -p "gi|568815592:29791752-29792749" > hla_variants.vcfVariants can also be inferred strictly from topology by not using -e, though unlike the above example, cycles are not supported. "Deconstruct" the VCF variants that were used to construct the graph. The output will be similar but identical to small/x.vcf.gz as vg construct can add edges between adjacent alts and/or do some normalization:
# using the same graph from the `map` example
vg deconstruct x.xg > x.vcfAs with vg call, it is best to compute snarls separately and pass them in with -r when working with large graphs.
A variety of commands are available:
- construct: graph construction
- view: conversion (dot/protobuf/json/GFA)
- index: index features of the graph in a disk-backed key/value store
- find: use an index to find nodes, edges, kmers, or positions
- paths: traverse paths in the graph
- align: local alignment
- map: global alignment (kmer-driven)
- stats: metrics describing graph properties
- join: combine graphs (parallel)
- concat: combine graphs (serial)
- ids: id manipulation
- kmers: generate kmers from a graph
- sim: simulate reads by walking paths in the graph
- mod: various transformations of the graph
- surject: force graph alignments into a linear reference space
- msga: construct a graph from an assembly of multiple sequences
- validate: determine if graph is valid
- filter: filter reads out of an alignment
- augment: adds variation from aligned reads into the graph
- call/genotype: call variants from an augmented graph
vg is a collection of tools based on a common data model (the variation graph) that is described by a protobuf schema (vg.proto). Data objects defined in vg.proto may be serialized via a stream pattern defined in stream.hpp. It is not necessary to write code in vg in order to interface with the algorithms defined here. Rather, it is sometimes simpler to write an external algorithm that reads and writes the same data formats.
MIT