This is the comparative genomics pipeline for PathoSPOT, the Pathogen Sequencing Phylogenomic Outbreak Toolkit.
The pipeline is run on sequenced pathogen genomes, for which metadata (dates, locations, etc.) are kept in a relational database (either SQLite or MySQL), and it produces output files that can be interactively visualized with pathoSPOT-visualize.
![]()
For example output and a live demo, please see the PathoSPOT website. Below, we provide documentation on how to setup and run the pipeline on your own computing environment. If you use this software, please cite our paper:
Berbel Caban A, Pak TR, Obla A et al. PathoSPOT genomic epidemiology reveals under-the-radar nosocomial outbreaks. Genome Medicine 2020 Nov 16;12(1):96. PMID:33198787 doi:10.1186/s13073-020-00798-3
This pipeline runs on Linux; however, Mac and Windows users can use Vagrant to rapidly build and launch a Linux virtual machine with the pipeline ready-to-use, either locally or on cloud providers (e.g., AWS). This bioinformatics pipeline requires ruby 2.2 or 2.3 with rake ≥12.3.3 and bundler, python 2.7 with the modules in requirements.txt, MUMmer 3.23, the standard Linux build toolchain, and additional software that the pipeline will build and install itself.
Download and install Vagrant using any of the official installers for Mac, Windows, or Linux. Vagrant supports both local virtualization via VirtualBox and cloud hosts (e.g., AWS).
The fastest way to get started with Vagrant is to install VirtualBox. Then, clone this repository to a directory, cd into it, and run the following:
$ vagrant up
It will take a few minutes for Vagrant to download a vanilla Debian 9 "Stretch" VM and configure it. Once it's done, to use your new VM, type
$ vagrant ssh
You should see the bash prompt vagrant@stretch:/vagrant$, and may proceed to Usage below.
The next time you want to use the pipeline in this VM, you won't need to start all over again; simply logout of your VM and vagrant suspend to save its state, and vagrant resume; vagrant ssh to pick up where you left off.
Vagrant can also run this pipeline on the AWS cloud using your AWS credentials. See README-vagrant-aws.md.
Mount Sinai users getting started on the Minerva computing environment can use an included script to setup an appropriate environment on a Chimera node (Vagrant is unnecessary); for more information see README-minerva.md.
You may be able to install prerequisites directly on a Linux machine by editing scripts/bootstrap.debian-stretch.sh to fit your distro's needs. As the name suggests, this script was designed for Debian 9 "Stretch", but will likely run with minor changes on most Debian-based distros, including Ubuntu and Mint. Note that this script must be run as root, expects the pipeline will be run by $DEFAULT_USER i.e. UID=1000, and assumes this repo is already checked out into /vagrant.
Rake, aka Ruby Make, is used to kick off the pipeline. Some tasks require certain parameters, which are provided as environment variables (and detailed more below). A quick primer on how to use Rake:
$ rake -T # list the available tasks
$ rake $TASK_1 $TASK_2 # run the tasks named $TASK_1 and $TASK_2
$ FOO="bar" rake $TASK_1 # run $TASK_1 with variable FOO set to "bar"
Important: If you are not using Vagrant, whenever firing up the pipeline in a new shell, you must always run source scripts/env.sh before running rake. The Vagrant environment does this automatically via ~/.profile.
If you used Vagrant to get started, it automatically downloads the example dataset (tar.gz) for MRSA isolates at Mount Sinai. The genomes are saved at example/igb and their metadata is in example/mrsa.db. (If you are not using Vagrant, you can run rake example_data to do the same.) Default environment variables in scripts/env.sh are configured so that the pipeline will run on the example data.
To run the full analysis, run the following, which invokes the three main tasks (parsnp, epi, and encounters, explained more below).
$ rake all
When the analysis finishes, there will be four output files saved into out/, which include a YYYY-MM-DD formatted date in the filename and have the following extensions:
.parsnp.heatmap.json→ made byparsnp; contains the genomic SNP distance matrix.parsnp.vcfs.npz→ made byparsnp; contains SNP variant data for each genome.encounters.tsv→ made byencounters; contains spatiotemporal data for patients.epi.heatmap.json→ made byepi; contains culture test data (positives and negatives)
These outputs can be visualized using pathoSPOT-visualize, which the Vagrant environment automatically installs and sets up for you. If you used VirtualBox, simply go to http://localhost:8989, which forwards to the virtual machine. For AWS, navigate instead to your public IPv4 address, which you can obtain by running the following within the EC2 instance:
$ curl http://169.254.169.254/latest/meta-data/public-ipv4; echo
By default, all output for tasks is saved in out/. To change this, set the OUT environment variable.
rake parsnp uses Parsnp from HarvestTools to create intraspecific genome alignments (on assembly sequences in FASTA files, with optional gene annotations in BED format). An optional (but recommended) preclustering step is performed with Mash to only align clusters of genomes that appear closely related, allowing these alignments to include a larger core genome and increase confidence that SNP counts will accurately reflect genetic divergence.

This task requires you to set the IGB_DIR, PATHOGENDB_URI, and IN_QUERY environment variables. When using the example environment, these variables are set for you and run a full analysis on the example dataset.
IGB_DIR: The full path to a directory containing the genome assemblies, in FASTA format. Each of these files should be in its own subdirectory named identically minus the.faor.fastaextension. Each subdirectory may also contain a BED file with gene annotations. See theigbdirectory in the example dataset (tar.gz).PATHOGENDB_URI: A URI to the database containing metadata on the genome assemblies; for SQLite, it issqlite://followed by a relative path to the file, and for MySQL the format ismysql2://user:password@host/db_name. See README-database.md to learn how to build your own database.IN_QUERY: AnSQL WHEREclause that can filter which assemblies in the database are included in the analysis. For our example,1=1is used, which simply uses all of the assemblies. For your databases you create, it will likely become useful to filter by species and/or location. The query can include any of the columns in thetAssemblies,tExtracts,tStocks,tIsolates,tOrganismsandtHospitalstables.
You may optionally specify two additional environment variables MASH_CUTOFF and MAX_CLUSTER_SIZE that tune the Mash preclustering step. To disable Mash preclustering, set both of these to the value 0. There is a third optional variable that disables recombination filtering.
MASH_CUTOFF: The maximum diameter, in Mash units, of each cluster. Mash units approximate average nucleotide identity (ANI). The default is 0.02, approximating 98% ANI (1 - 0.02) among all genomes in each cluster.MAX_CLUSTER_SIZE: The maximum number of assemblies to allow in each cluster before forcing a split. The default is 100. This should be greater than the largest conceivable outbreak you could expect in your dataset. If the heatmap in pathoSPOT-visualize warns you about this, we recommend rerunning with a higher number to see if your outbreak clusters grow larger.DISABLE_PHIPACK: By default, this task will configure parsnp to use PhiPack to filter SNPs in likely regions of recombination. Set this variable to anything to disable this behavior.
This tasks creates two final output files which include a YYYY-MM-DD formatted date in the filename and have the following extensions:
.parsnp.heatmap.json→ contains the genomic SNP distance matrix and other metadata, in JSON format.parsnp.vcfs.npz→ contains SNP variant data for each genome, in NumPy NPZ format.

When these are placed in the data/ directory of pathoSPOT-visualize, it enables the "heatmap" visualization as seen above.
rake encounters produces a TSV file containing all the spatiotemporal data for patients associated with particular pathogen genomes. It requires the same three arguments as rake parsnp: IGB_DIR, PATHOGENDB_URI, and IN_QUERY. Note that IN_QUERY is still principally filtering assemblies; all patients associated with those assemblies are included in the TSV output.
The output file includes a YYYY-MM-DD formatted date in the filename and ends with .encounters.tsv.
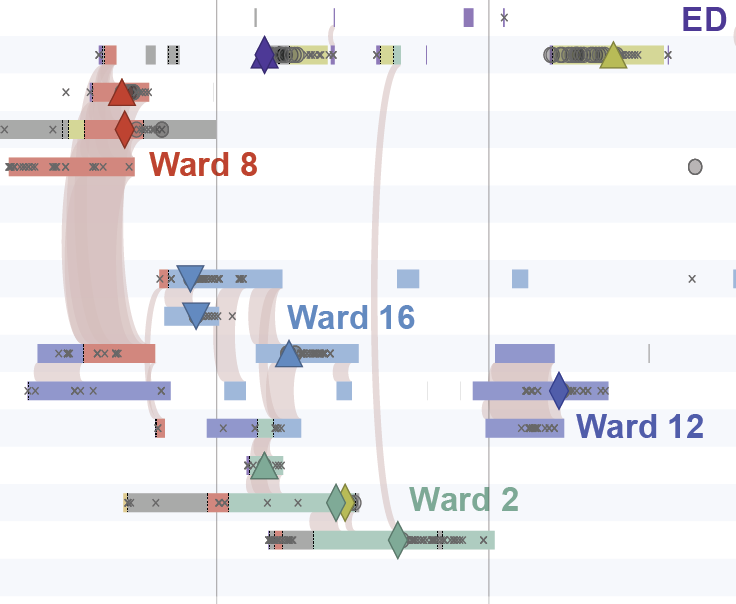
When provided to pathoSPOT-visualize alongside the outputs from rake parsnp, it enables the "timeline" visualization within the "dendro-timeline", as seen above.
rake epi produces a JSON file containing spatiotemporal data for all isolates and culture tests (both positive and negative) for the patients associated with particular pathogen genomes. It requires the same three arguments as rake parsnp: IGB_DIR, PATHOGENDB_URI, and IN_QUERY.
The output file includes a YYYY-MM-DD formatted date in the filename and ends with .epi.heatmap.json.
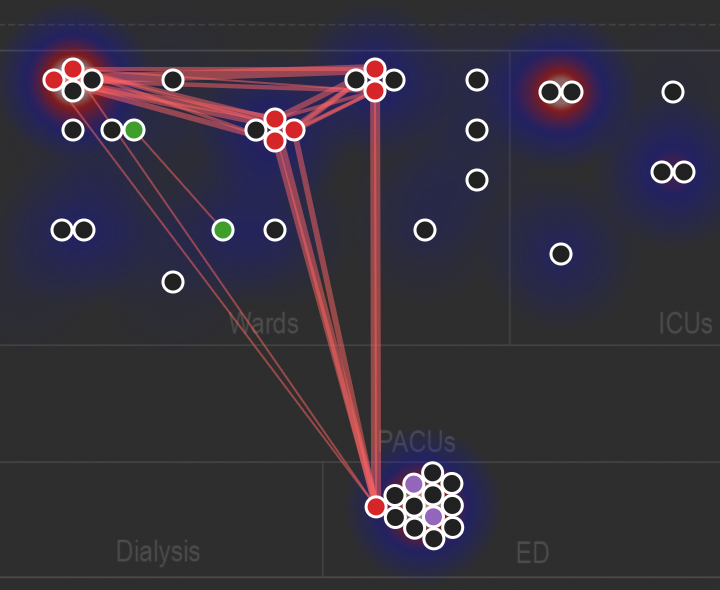
When provided to pathoSPOT-visualize alongside the outputs from rake parsnp, it enables the display of non-sequenced culture results in the "dendro-timeline" as well as the density map of collected isolates underlying the "network" visualization, seen above.
This is a shortcut for rake parsnp encounters epi, which runs all three of those tasks with the same environment variables.
This downloads the example dataset (tar.gz) into example/, if it is not already present.
Besides the genome sequences and their gene annotations, the pipeline expects a metadata database supplied via the PATHOGENDB_URI parameter. We provide an example database called mrsa.db in the example dataset (tar.gz), which you can open and modify to begin analyzing your own specimens. You do not need to be a programmer or pay for software to do so; we used SQLite which is free and open-source software with many available GUI tools.
To learn how to get started on your own database, see README-database.md.
If you want to copy the final outputs outside of the Vagrant environment, e.g. to serve them with pathoSPOT-visualize from a different machine, use vagrant-scp as follows from the host machine:
$ vagrant plugin install vagrant-scp
$ vagrant scp default:/vagrant/out/*.json /destination/on/host
$ vagrant scp default:/vagrant/out/*.npz /destination/on/host
$ vagrant scp default:/vagrant/out/*.encounters.tsv /destination/on/host
This pipeline downloads and installs the appropriate versions of Mash and HarvestTools into vendor/.
Software in this repository (not inclusive of library dependencies or software downloaded and installed alongside it when it is executed) is licensed under the standard MIT license, a permissive free software license. See the LICENSE.txt for more details.