![]()


Protein-protein interaction (PPI) networks are a fundamental resource for modeling cellular and molecular function, and a large and sophisticated toolbox has been developed to leverage their structure and topological organization to predict the functional roles of under-studied genes, proteins, and pathways. However, the overwhelming majority of experimentally-determined interactions from which such networks are constructed come from a small number of well-studied model organisms. Indeed, most species lack even a single experimentally-determined interaction in these databases, much less a network to enable the analysis of cellular function, and methods for computational PPI prediction are too noisy to apply directly. We introduce PHILHARMONIC, a novel computational approach that couples deep learning de novo network inference (D-SCRIPT) with robust unsupervised spectral clustering algorithms (Diffusion State Distance) to uncover functional relationships and high-level organization in non-model organisms. Our clustering approach allows us to de-noise the predicted network, producing highly informative functional modules. We also develop a novel algorithm called ReCIPE, which aims to reconnect disconnected clusters, increasing functional enrichment and biological interpretability. We perform remote homology-based functional annotation by leveraging hmmscan and GODomainMiner to assign initial functions to proteins at large evolutionary distances. Our clusters enable us to newly assign functions to uncharacterized proteins through "function by association." We demonstrate the ability of PHILHARMONIC to recover clusters with significant functional coherence in the reef-building coral P. damicornis, its algal symbiont C. goreaui, and the well-annotated fruit fly D. melanogaster. We perform a deeper analysis of the P. damicornis network, where we show that PHILHARMONIC clusters correlate strongly with gene co-expression and investigate several clusters that participate in temperature regulation in the coral, including the first putative functional annotation of several previously uncharacterized proteins. Easy to run end-to-end and requiring only a sequenced proteome, PHILHARMONIC is an engine for biological hypothesis generation and discovery in non-model organisms.
- Installation
- Usage
- Workflow Overview
- Interpreting Results
- Detailed Configuration
- Citation
- FAQ/Known Issues
- Contributing
pip install philharmonicWe also recommend installing Cytoscape to visualizing the resulting networks.
The only data that PHILHARMONIC requires is a set of protein sequences in .fasta format. We provide a set of high-level GO terms on which to filter proteins prior to candidate generation and network prediction. You may optionally provide your own set of GO terms, as the go_filter_path argument in the configuration file.
The config.yml file is where you will specify the parameters for PHILHARMONIC. We provide a sample config in this repository
with recommended parameters. You will need to specify the paths to your protein sequences. You can find an explanation for all parameters below. If you want to use an LLM to automatically name your clusters, make sure you have set the OPENAI_API_KEY environment variable with your API key, or set llm.model in the config to an open source LLM (see llm package docs). If you use a different configuration file name or location, you can specify it with the --configfile flag when running Snakemake.
# User Specified
run_name: [identifier for this run]
sequence_path: [path to protein sequences in .fasta format]
work_dir: [path to working directory]
use_llm: [true/false: whether to name clusters using a large language model]
...Once your configuration file is set up, you can invoke PHILHARMONIC with
philharmonic conduct -cf {config file} -c {number of cores}We provide a zip of the most relevant output files in [run].zip, which contains the following files:
run.zip
|
|-- run_human_readable.txt # Easily readable/scannable list of clusters
|-- run_network.positive.tsv # All edges predicted by D-SCRIPT
|-- run_clusters.json # Main result file, contains all clusters, edges, and functions
|-- run_cluster_graph.tsv # Graph of clusters, where edges are weighted by the number of connections between clusters
|-- run_cluster_graph_functions.tsv # Table of high-level cluster functions from GO Slim
|-- run_GO_map.tsv # Mapping between proteins and GO function labelsInstructions for working with and evaluating these results can be found in Interpreting the Results.
We provide support for running PHILHARMONIC in Google Colab with the notebook at nb/00_run_philharmonic.ipynb. However, we note that the hmmscan and dscript sections can be quite resource intensive, and may result in a time-out if run on Colab.
A detailed overview of PHILHARMNONIC can be found in the manuscript. We briefly outline the method below.
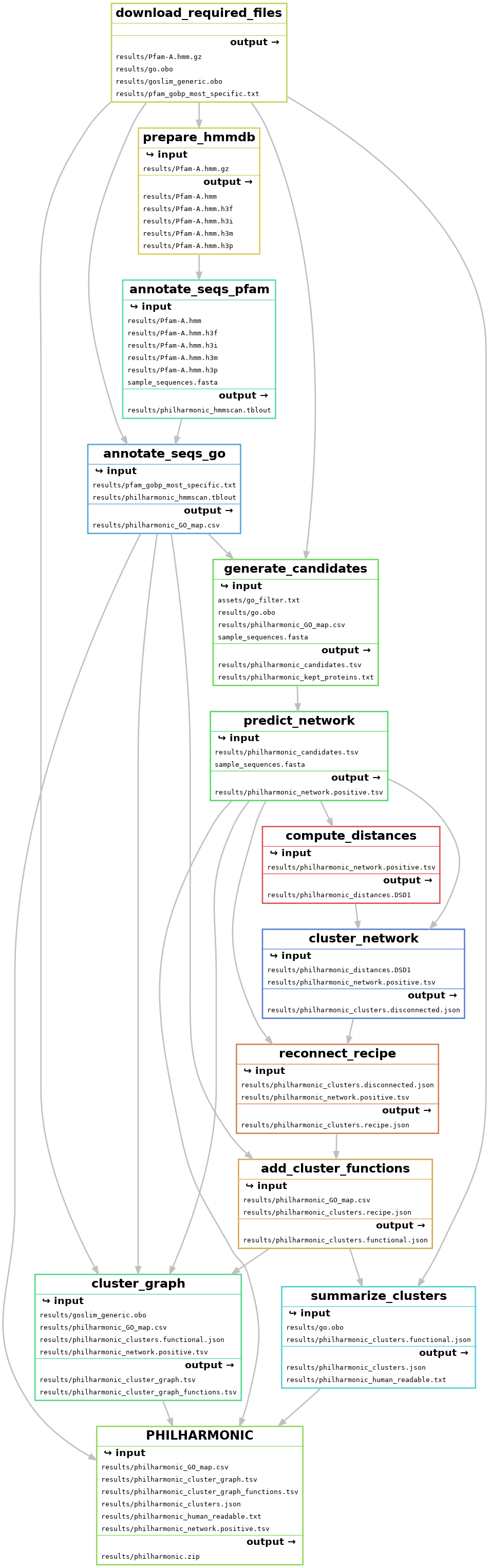
- Download necessary files (
download_required_files) - Run hmmscan on protein sequences to annotate pfam domains (
annotate_seqs_pfam) - Use pfam-go associations to add GO terms to sequences (
annotate_seqs_go) - Generate candidate pairs (
generate_candidates+) - Use D-SCRIPT to predict network (
predict_network) - Compute node distances with FastDSD (
compute_distances) - Cluster the network with spectral clustering (
cluster_network+) - Use ReCIPE to reconnect clusters (
reconnect_recipe) - Annotate clusters with functions (
add_cluster_functions+) - Compute cluster graph (
cluster_graph+) - Name and describe clusters for human readability (
summarize_clusters+)
Each of these steps can be invoked independently by running snakemake -c {number of cores} --configfile {config file} {target}. The {target} is shown in parentheses following each step above. Certain steps (marked with a +) are available to run directly as philharmonic commands with the appropriate input, e.g. philharmonic summarize-clusters---note that in this case, underscores are generally replaced with dashes. Run philharmonic --help for full specifications.
We provide some guidance on interpreting the output of PHILHARMONIC here, as well as analysis notebooks which can be run locally or in Google Colab. The typical starting point for these analyses is the zip file described in Outputs.
Using the clusters.json file, the network.positive.tsv file, the GO map.tsv file, and a GO Slim database, you can view the overall network, a summary of the clustering, and explore individual clusters.
| Network | |
|---|---|
| Nodes | 7267 |
| Edges | 348278 |
| Degree (Med) | 37 |
| Degree (Avg) | 95.8519 |
| Sparsity | 0.00659501 |
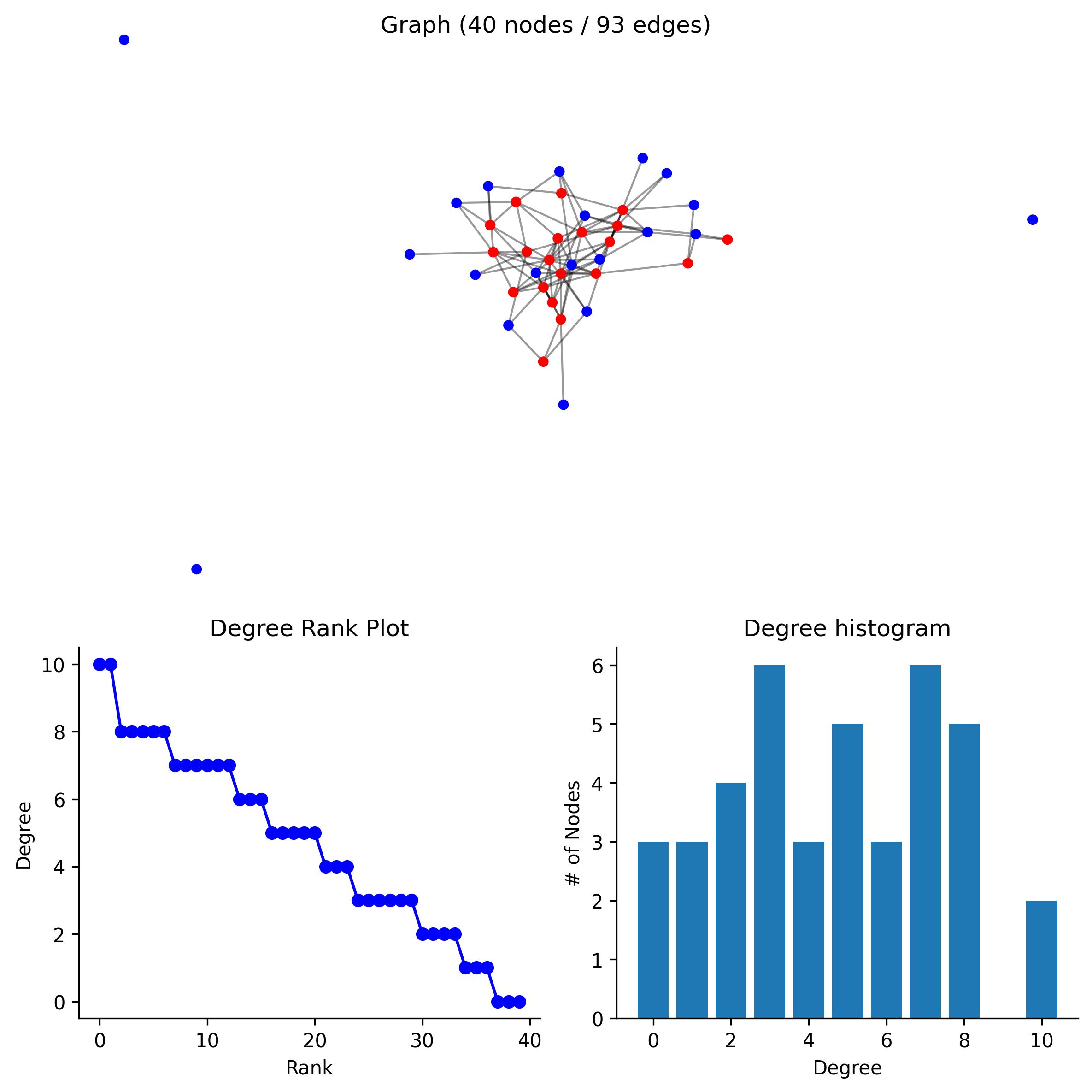
Pain Response and Signaling Pathways Cluster
Cluster of 20 proteins [pdam_00013683-RA, pdam_00006515-RA, pdam_00000216-RA, ...] (hash 208641124039621440)
20 proteins re-added by ReCIPE (degree, 0.75)
Edges: 3
Triangles: 0
Max Degree: 2
Top Terms:
GO:0019233 - <sensory perception of pain> (20)
GO:0048148 - <behavioral response to cocaine> (19)
GO:0006468 - <protein phosphorylation> (19)
GO:0007507 - <heart development> (19)
GO:0010759 - <positive regulation of macrophage chemotaxis> (19)
GO:0001963 - <synaptic transmission, dopaminergic> (19)
GO:0071380 - <cellular response to prostaglandin E stimulus> (19)
GO:0071502 - <cellular response to temperature stimulus> (19)
GO:0008542 - <visual learning> (19)
GO:0007601 - <visual perception> (19)Using the same files, you can run a statistical test of cluster function by permuting cluster labels, and computing the Jaccard similarity between terms in the same cluster.

You can view GO enrichments for each cluster using g:Profiler. In the provided notebook, we perform an additional mapping step to align the namespace used in our analysis with the namespace used by g:Profiler.
| native | name | p_value | |
|---|---|---|---|
| 0 | GO:0007186 | G protein-coupled receptor signaling pathway | 4.99706e-09 |
| 1 | GO:0007165 | signal transduction | 2.77627e-06 |
| 2 | GO:0023052 | signaling | 3.17572e-06 |
| 3 | GO:0007154 | cell communication | 3.50392e-06 |
| 4 | GO:0051716 | cellular response to stimulus | 1.58692e-05 |
| 5 | GO:0050896 | response to stimulus | 2.62309e-05 |
| 6 | GO:0050794 | regulation of cellular process | 0.000432968 |
| 7 | GO:0050789 | regulation of biological process | 0.00072382 |
| 8 | GO:0065007 | biological regulation | 0.000923115 |
If gene expression data is available for the target species, we can check that proteins clustered together have correlated expression, and we can visualize where differentially expressed genes localize within the networks and clusters. Here, we use Pocillopora transcriptomic data from Connelly et al. 2022.

- Load
network.positive.tsvusingFile -> Import -> Network from File
- Load
cluster_graph.tsvusingFile -> Import -> Network from File - Load
cluster_graph_functions.tsvusingFile -> Import -> Table from File - Add a
Column filteron theEdge: weightattribute, selecting edges greater than ~50-100 weight Select -> Nodes -> Nodes Connected by Selected Edgesto subset the nodes- Create the subgraph with
File -> New Network -> From Selected Nodes, Selected Edges - Layout the network with your layout of choice, we recommend
Layout -> Prefuse Force Directed Layout -> weight - Add node colors using the PHILHARMONIC style, imported with
File -> Import -> Styles from File

The config.yml file contains various parameters that control the behavior of PHILHARMONIC. Below is a detailed explanation of each parameter, including default values:
run_name: Identifier for this run [required]sequence_path: Path to protein sequences in .fasta format [required]go_filter_path: Path to list of GO terms to filter candidates (default: "assets/go_filter.txt")work_dir: Path to the working directory where results will be stored (default: "results")use_llm: Boolean flag to enable/disable LLM naming for cluster summarization (default: true)
Note: if you set use_llm with an OpenAI model, make sure that you have set the environment variable OPENAI_API_KEY prior to running.
seed: Random seed for reproducibility (default: 42)
hmmscan.path: Path to the hmmscan executable (default: "hmmscan")hmmscan.threads: Number of threads to use for hmmscan (default: 32)
dscript.path: Path to the D-SCRIPT executable (default: "dscript")dscript.n_pairs: Number of protein pairs to predict (-1 for all pairs) (default: -1)dscript.model: Pre-trained D-SCRIPT model to use. (default: "samsl/dscript_human_v1")dscript.device: GPU device to use (-1 for CPU) (default: 0)
dsd.path: Path to the FastDSD executable (default: "fastdsd")dsd.t: Edge existence threshold for DSD algorithm (default: 0.5)dsd.confidence: Boolean flag to use confidence scores (default: true)
clustering.init_k: Initial number of clusters for spectral clustering (default: 500)clustering.min_cluster_size: Minimum size of a cluster (default: 3)clustering.cluster_divisor: Divisor used to determine the final number of clusters (default: 20)clustering.sparsity_thresh: Sparsity threshold for filtering edges (default: 1e-5)
recipe.lr: Linear ratio for ReCIPE algorithm (default: 0.1)recipe.cthresh: Connectivity threshold to add proteins until for ReCIPE (default: 0.75)recipe.max_proteins: Maximum number of proteins to add to a cluster in ReCIPE (default: 20)recipe.metric: Metric to use for ReCIPE (default: "degree")
llm.model: Language model to use for cluster summarization (default: "gpt-4o")
@article{sledzieski2024decoding,
title={Decoding the Functional Interactome of Non-Model Organisms with PHILHARMONIC},
author={Sledzieski, Samuel and Versavel, Charlotte and Singh, Rohit and Ocitti, Faith and Devkota, Kapil and Kumar, Lokender and Shpilker, Polina and Roger, Liza and Yang, Jinkyu and Lewinski, Nastassja and Putnam, Hollie and Berger, Bonnie and Klein-Seetharaman Judith and Cowen, Lenore},
journal={BioRxiv},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}- On Linux, the package
placmay not install properly with the includedenvironment.yml. If you are seeing the errorNo module names 'asyncore', try runningmamba update plac
mamba create -n philharmonic python==3.11
pip install poetry
git clone https://github.com/samsledje/philharmonic.git
cd philharmonic
poetry install
pre-commit install
git checkout -b [feature branch]