This is a repository for a source code to replicate Sakaue S et al. Nature Communications. 2020.
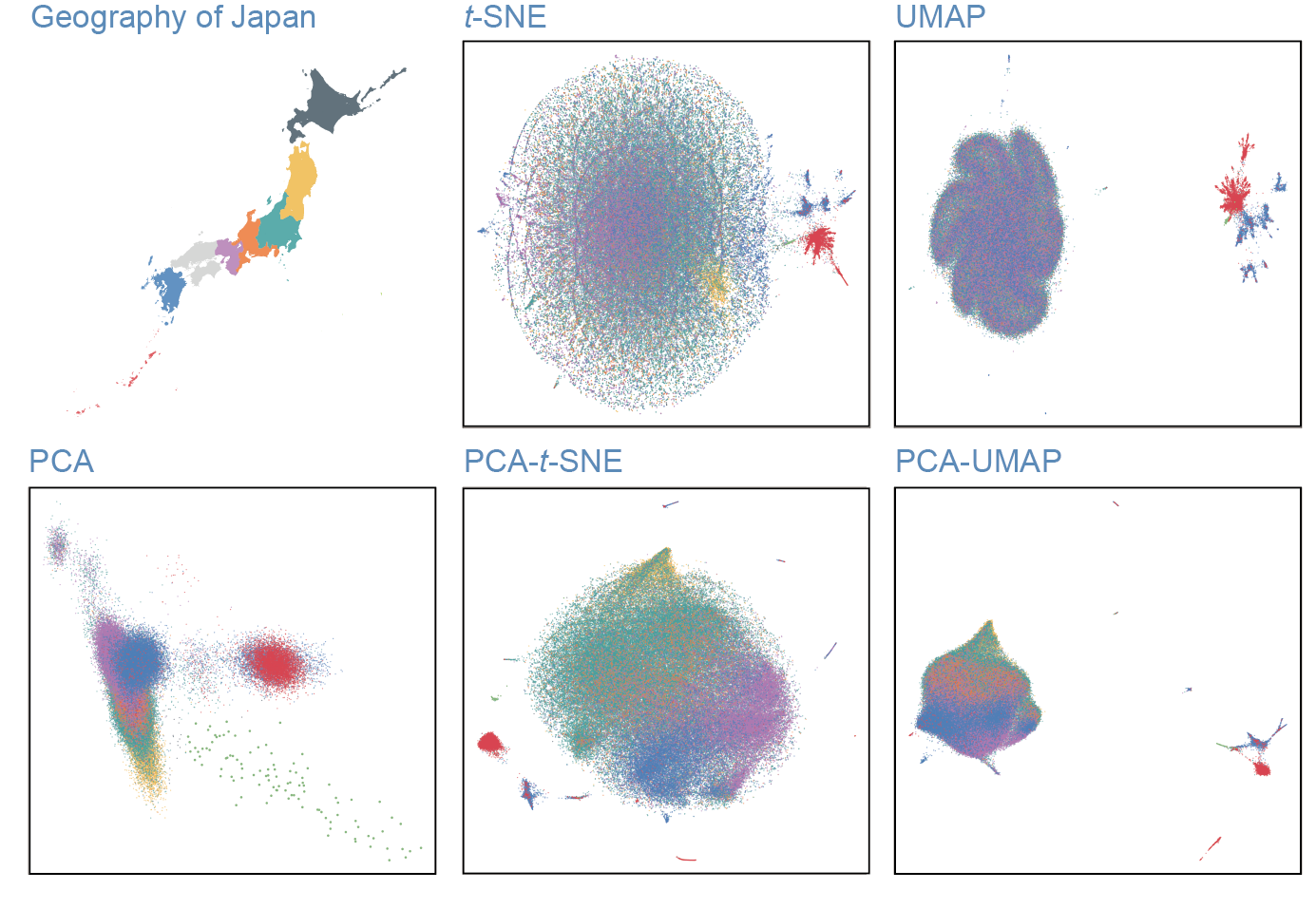
To find finest-scale population substructures, we applied a series of dimensionality reduction methods to large-scale genotype data of Japanese and also worldwide populations. We will introduce the python-based source code for replicating our results.
Our paper is out!
Sakaue S et al. Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction. Nature Communications. 2020
- umap-learn
- scikit-learn
- scipy
- pandas
- MulticoreTSNE
We would recommend you to install these dependencies by using anaconda.
In addition, plink version 1.9 (https://www.cog-genomics.org/plink2/) should be in your $PATH.
If you would like to download the scripts,
$ git clone https://github.com/saorisakaue/Genotype-dimensionality-reduction
$ cd ./Genotype-dimensionality-reduction
User input is genotype data in plink format.
plinkfile_prefix.{bed/bim/fam}
Important!
To run dimensionality reduction methods effectively, we would recommend to impute all missing genotypes in your plink file.
Such imputation tools are;
Beagle(https://faculty.washington.edu/browning/beagle/beagle.html)Eagle(https://data.broadinstitute.org/alkesgroup/Eagle/)
Eagle is scalable and accurate for imputing biobank-scale genotype data (e.g., N > 100K).
Two scripts
01_prep_genotype.sh: for LD pruning and formatting of the genotype02_run_dr.py: main dimensionality reduction functions
are used for performing dimensionality reduction methods on genotype data provided by the user as plink file. To run the script on the command line, simply
$ bash 01_prep_genotype.sh ${path/to/plinkfile_prefix}
$ python 02_run_dr.py ${path/to/plinkfile_prefix}
can do all the analyses.
Example genotype data of 50 individuals from 1KGPp3v5 is provided in ./data_umap directory.
If you use example dataset, the command line will be as follows;
$ bash 01_prep_genotype.sh ./data_umap/1KG.selected
$ python 02_run_dr.py ./data_umap/1KG.selected
Please note that it would be efficient to split the process within 02_run_dr.py into each part of the five dimensionality reduction methods, particularly when the genotype data is large (N > 10K).
Any questions? Saori Sakaue (ssakaue[at]sg.med.osaka-u.ac.jp)