![]()




- Song W, Steensen K, Thomas T. (2017) HgtSIM: a simulator for horizontal gene transfer (HGT) in microbial communities. PeerJ 5:e4015 https://doi.org/10.7717/peerj.4015 PDF
- Contact: Weizhi Song (songwz03@gmail.com), Torsten Thomas(t.thomas@unsw.edu.au)
- Affiliation: The Centre for Marine Bio-Innovation (CMB), The University of New South Wales, Sydney, Australia

-
2019-01-06:
- HgtSIM can be installed with "pip3 install HgtSIM" now.
-
2018-04-06:
- combined the '-mixed', '-mini' and '-maxi' options into one: '-mixed min-max'.
-
2017-09-16:
- add support for draft genome.
- add support for dynamic flanking sequences.
- add support for the 'mixed' mode.
- add support for the 'keep_cds' option.
- run Prodigal if "-keep_cds" was specified
- check Ns in provided gene sequences
- check whether provided sequences to transfer are ORFs, exit if not
-
HgtSIM is implemented in python3, you can install it with:
pip3 install HgtSIM -
HgtSIM requires BLAST+, you can either add it to your system path or specify full path to "blastn" and "blastp" executables with options "-blastn" and "-blastp".
HgtSIM -h
-t sequences of genes to be transferred (multi-fasta format)
-i mutation level
-d distribution of transfers to the recipient genomes
-f folder holds recipient genomes
-r ratio of mutation types
-x file extension of recipient genomes
-lf left end flanking sequences
-rf right end flanking sequences
-mixed randomly assign mutation levels between specified values, parameter format: min-max
-keep_cds insert transfers only to non-coding regions, need the annotation files (in gbk format) of recipient genomes
-a folder holds the annotation files (in gbk format) of recipient genomes
-l minimum length of intergenic region to be considered for insertion
-blastn path to blastn executable, default: blastn
-blastp path to blastp executable, default: blastp
-
Sequences of genes to be transferred (in multi-fasta format).
-
A folder holds all recipient genomes, one file per genome.
-
The mutation level of genes to be transferred. This can be specified either as a fixed value, or within a range (the 'mixed' mode). If the 'mixed' argument was provided, HgtSIM will randomly select a value between user specified minimum and maximum mutation levels to alter each gene transfer.
# with fixed mutation level (e.g. 10%). HgtSIM -t genes.fasta -d distribution.txt -f input_genomes -r 1-0-1-1 -x fna -i 10 # with 'mixed' mode (e.g. 5-25%) HgtSIM -t genes.fasta -d distribution.txt -f input_genomes -r 1-0-1-1 -x fna -mixed 5-25 -
The ratio of mutation categories (separated with dash). The default setting is '1-0-1-1'. Please refer to the publication (http://dx.doi.org/10.7717/peerj.4015) or the figure below for its setting.
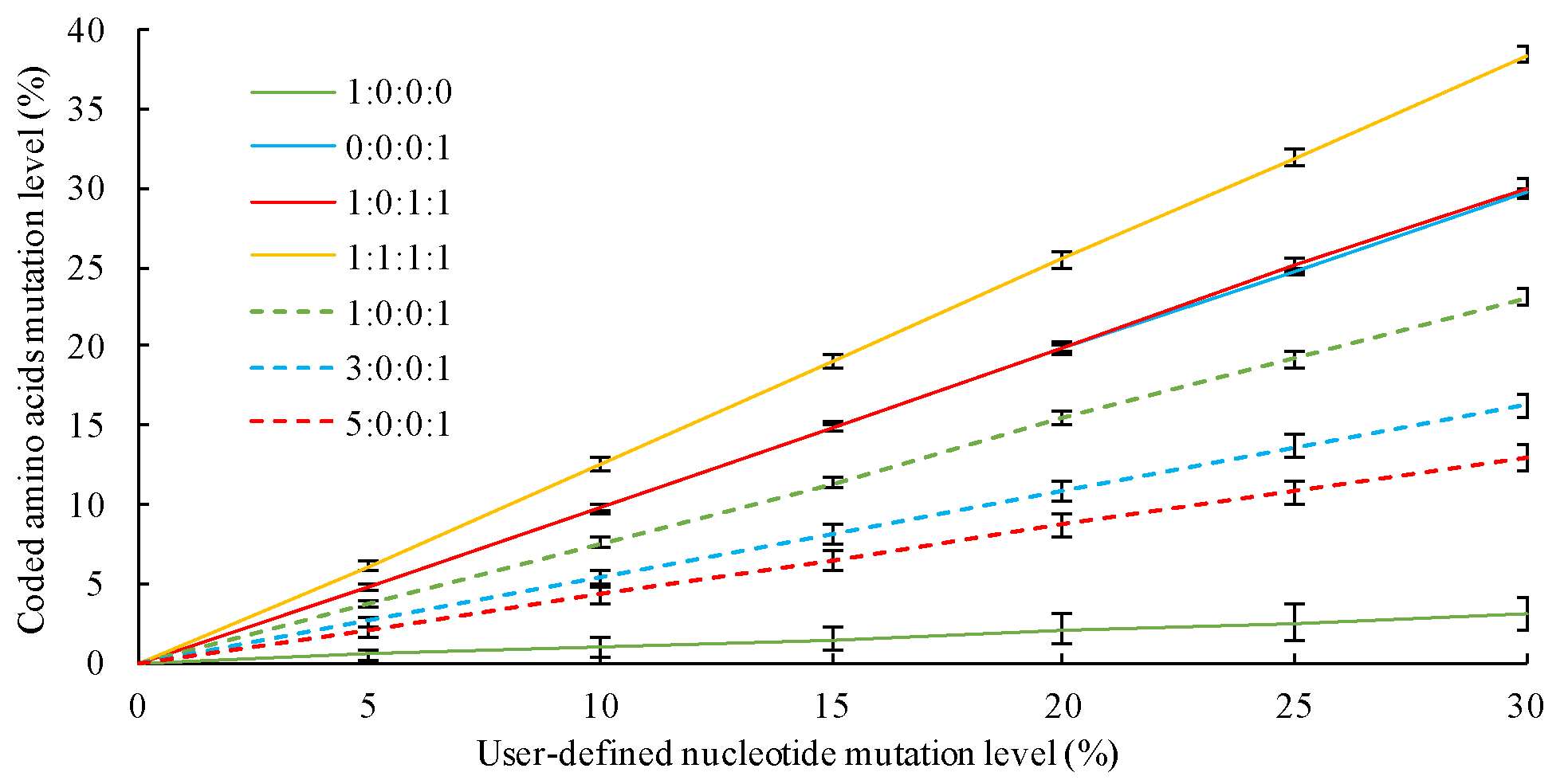
-
The distribution of transfers to the recipient genomes. The first column refers to the recipient genomes(without file extension), followed by a list of genes to be transferred therein (separated with comma).
BAD,AAM_03063,AKV_01007,AMAC_01196,AMAU_02632,AMS_01785 BDS,AAM_00175,AKV_00943,AMAC_00215,AMAU_02085,AMS_01465 BGC,AAM_00176,AKV_01272,AMAC_01576,AMAU_00617,AMS_02653 BHS,AAM_00195,AKV_01273,AMAC_01674,AMAU_05963,AMS_03303 BNM,AAM_00209,AKV_00282,AMAC_02914,AMAU_02414,AMS_03378 BRT,AAM_00308,AKV_02353,AMAC_03303,AMAU_00830,AMS_01655 -
The flanking sequences to be added to the end of gene transfers. Can be specified with '-lf' and '-rf', the default value is None.
# introduce gene transfers without adding flanking sequences HgtSIM -t genes.fasta -i 10 -d distribution.txt -f input_genomes -r 1-0-1-1 -x fna # or, add same pair of flanking sequences (e.g. 'TAGATGAGTGATTAGTTAGTTA') to all gene transfers HgtSIM -t genes.fasta -i 10 -d distribution.txt -f input_genomes -r 1-0-1-1 -x fna -lf TAGATGAGTGATTAGTTAGTTA -rf TAGATGAGTGATTAGTTAGTTA # or, add flanking sequences dynamically to the two ends of each gene transfer HgtSIM -t genes.fasta -i 10 -d distribution.txt -f input_genomes -r 1-0-1-1 -x fna -lf lf.fasta -rf rf.fastaif you want to add flanking sequences dynamically to the gene transfers, you can specify the left and right side sequences in two multi-fasta files. The IDs of the flanking sequences need to be exactly the same to their corresponding gene transfers.
As an illustration, if you have four transfers, which are transfer_A, transfer_B, transfer_C and transfer_D. And you have provided the following two files:
lf.fasta
>transfer_A AAAAAAAAAA >transfer_B TTTrf.fasta
>transfer_A GGGGGGG >transfer_C CCCCCHgtSIM will then:
- add 'AAAAAAAAAA' to the left and 'GGGGGGG' to the right end of transfer_A;
- add 'TTT' to the left and nothing to the right end of transfer_B;
- add nothing to the left and 'CCCCC' to the right end of transfer_C;
- add nothing to boths end of transfer_D.
-
Transfers can be inserted only to the intergenic regions by specifying the 'keep_cds' option. The annotation files (in genbank format) of the recipient genomes are needed to enable this option.

- Produced genomes with transferred genes, which were placed in folder 'Genomes_with_transfers'.
- The amino acid sequences of input genes to be transferred.
- The nucleotide and amino acid sequences of mutated input genes.
- The mutation report file, which includes two parts:
- on the top is the nc and aa identities between input and mutated sequences for each transfer.
- followed by a summary of changed nucleotide bases for each transfer.
- The insertion report file.