![]() TBSP: Trajectory Inference Based on SNP information.
TBSP: Trajectory Inference Based on SNP information.

-
python (python 2 and python 3 are both supported)
It was installed by default for most Linux distribution and MAC.
If not, please check https://www.python.org/downloads/ for installation instructions. -
Python packages dependencies:
-- scikit-learn
-- scipy
-- numpy
-- matplotlib
-- networkx
-- pyBigWig
-- Biopython -
other dependencies:
-- python-dev (python2) or python3-dev (python3)
It can be installed easily for most linux distributions. For example, debian/ubuntu:
sudo apt-get install python-dev
or
sudo apt-get install python3-dev
For Macos, it was installed by default.
The python setup.py script (or pip) will try to install these packages automatically. However, please install them manually if, by any reason, the automatic installation fails.
- Platform:
Macos and Linux verified. For windows, the dependent pyBigWig package is not available.
There are 3 options to install scdiff.
-
Option 1: Install from download directory
cd to the downloaded scdiff package root directory$cd tbsprun python setup to install
$python setup.py installMacOS or Linux users might need the sudo/root access to install. Users without the root access can install the package using the pip/easy_install with a --user parameter (install python libraries without root).
$sudo python setup.py installuse python3 instead of python in the above commands to install if using python3.
-
Option 2: Install from Github:
python 2:
$sudo pip install --trusted-host github.com --upgrade http://github.com/phoenixding/tbsp/zipball/masterpython 3:
$sudo pip3 install --trusted-host github.com --upgrade http://github.com/phoenixding/tbsp/zipball/master
The above pip installation options should be working for Linux and MacOS systems.
For MacOS users, it's recommended to use python3 installation. The default python2 in MacOS has
some compatibility issues with a few dependent libraries. The users would have to install their own
version of python2 (e.g. via Anocanda) if they prefer to use python2 in MacOS.
usage: tbsp [-h] -i IVCF [-b [IBW]] [-k KCLUSTER] [-l [CELL_LABEL]] -o
OUTPUT [--cutl CUTL] [--cuth CUTH] [--cutc CUTC]
optional arguments:
-h, --help show this help message and exit
-i IVCF, --ivcf IVCF Required,directory with all input .vcf files. This
specifies the directory of SNP files (.vcf) for the
cells (one .vcf file for each cell). These .vcf files
can be obtained using the provided bam2vcf script or
other RNA-seq variant calling pipelines preferred by
the users.
-b [IBW], --ibw [IBW]
Optional,directory with all input bigwig (.bw) files
with the information about the number of aligned reads
at each genomic position. These bigwig files are used
to filter the SNPs, which are redundant to expression
information.
-l [CELL_LABEL], --cell_label [CELL_LABEL]
Optional, labels for the cells. This is used only to
annotate the cells with known information, not used
for building the model.
-k KCLUSTER, --kcluster KCLUSTER
Optional, number of clusters, Integer. If not
specified, the program will choose the k with best
silhouette score.
-o OUTPUT, --output OUTPUT
Required,output directory
--cutl CUTL Optional, lower bound cutoff to remove potential false
positive SNPs, default=0.1
--cuth CUTH Optional, upper bound cutoff to remove baseline SNPs,
which are common in most cells, default=0.8
--cutc CUTC Optional, convergence cutoff, a smaller cutoff
represents a stricter convergence
criterion,default=0.001
-
-i:
Required input, this specifies the directory of all SNP(.vcf) files. We recommend using GATK RNA-seq variant calling pipeline to call the vcfs from .bam (mapped reads) files. Users are also allowed to use the SNPs (.vcfs) identified by programs of their preferences. -
-b:
Optional input, this specifies the directory of all bigwig (.bw) files. We provided the script bam2bw.py under bam2bw directory to convert the bam files to bigwig files. This files are used to filter SNPs, potentially redundant to expression. -
-l:
Optional input, this specifies the labels for the cells. File format (tab-delimited):
cell1 label1
cell2 label2
These cell labels are only used to annotate the cells in the trajectory. The other optional parameters are specified above.
-
GroupCells.txt:
A text file, which describes the cells in each cluster.Format:
Cell_ID Cluster_ID SRR1931024 cluter:0 SRR1930999 cluter:0 SRR1930977 cluter:0 SRR1931041 cluter:0 SRR1931012 cluter:0 SRR1930945 cluter:0 SRR1931003 cluter:0 SRR1931002 cluter:0 SRR1931004 cluter:0 .. -
SNP_matrix.tsv:
The SNP matrix for all the cells. Row: SNPs Column: Cells Value: Binary (0/1), which indicates whther the SNP is included in the cell. -
SNP_matrix.jpg:
The SNP matrix in jpg image.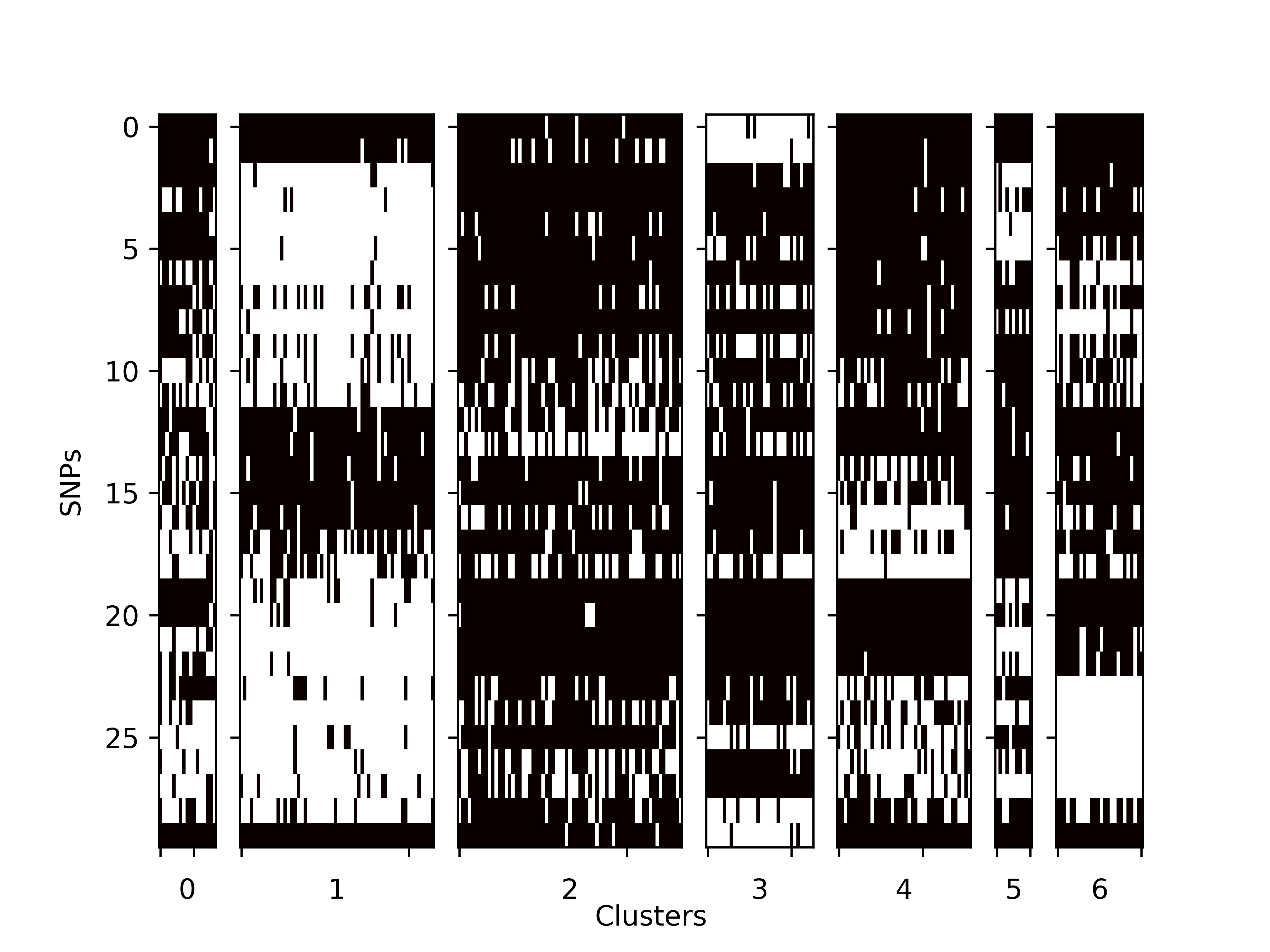
-
Trajectory.dat:
4 (-0.6168642633606496, -0.29504774213348317) Inner4 (-0.01784069226314263, -0.19237987266625067) 2 (-1.0, 0.1907479586411944) Inner5 (0.526511663382742, -0.048432121394020027) 6 (0.6167190582080249, -0.21090119687699874) 0 (-0.02807716265846153, -0.3564777531262085) 3 (-0.7661682030072015, 0.29873283057457906) Inner3 (-0.4986251141938113, -0.13657170781011707) Inner1 (0.785885623794461, 0.14046537917538365) 5 (0.8477737598817315, 0.3307492805568145) 1 (0.9509945159224894, 0.1308847244003101) Inner2 (-0.8003091857061821, 0.14823022065879607)First column: cluster id
second column: coordinates -
Trajectory.jpg:
Graph representation of Trajectory.dat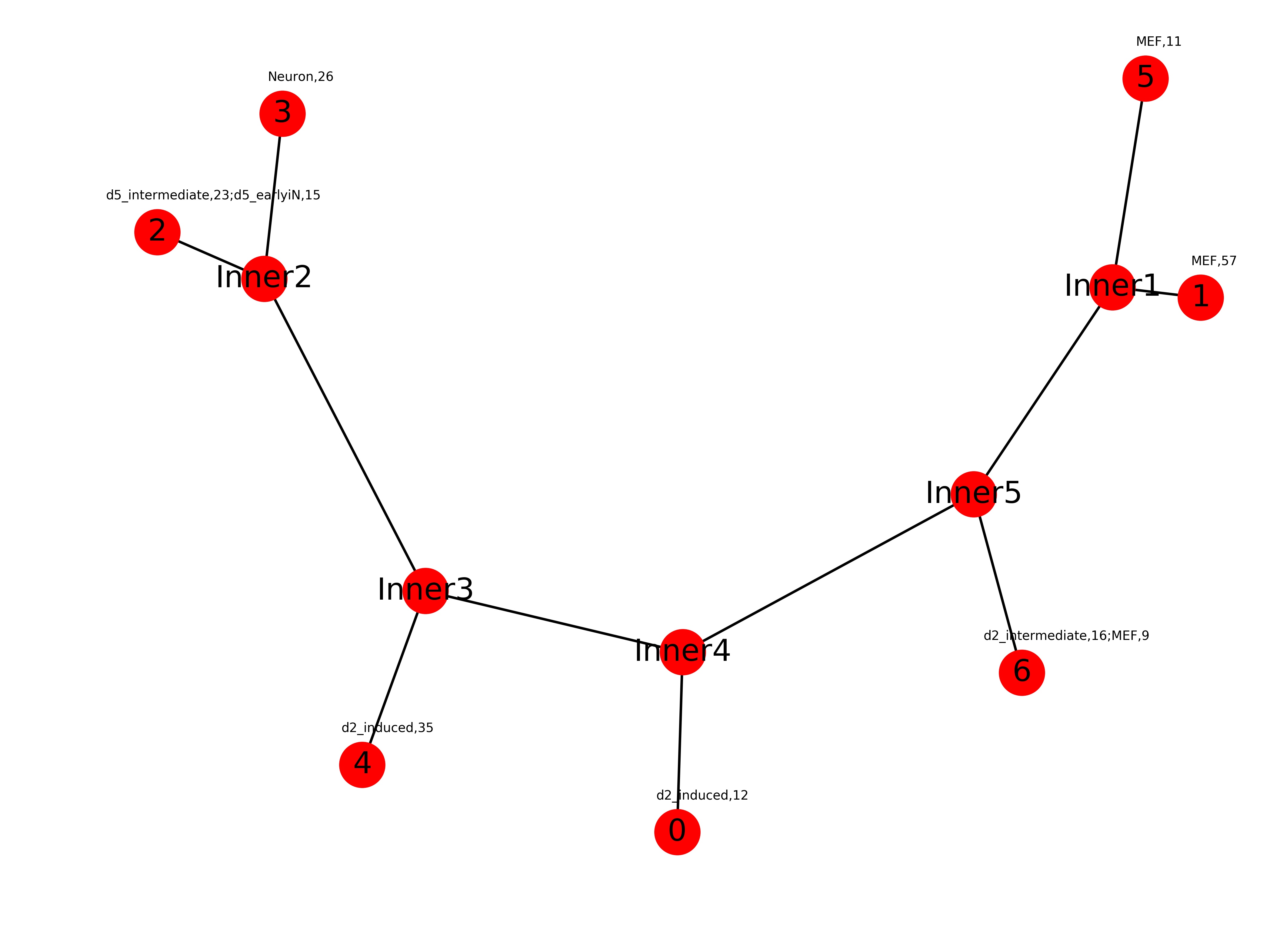


- Example inputs:
We provided example vcf files under examples folder. To run tbsp on the example data:
$tbsp -i examples/vcf_example -o example_out
- Example outputs:
Example output files can be found under examples folder.
This software was developed by ZIV-system biology group @ Carnegie Mellon University.
Implemented by Jun Ding.
This software is under MIT license.
zivbj at cs.cmu.edu
jund at cs.cmu.edu