
GeneBlocks is a Python library for comparing DNA sequences. It can be used to:
- Find common blocks in a group of DNA sequences, to factorize them (e.g. only analyze or synthetize each common block once).
- Highlight differences between sequences (insertions, deletions, mutations).
- Transfer Genbank features from one record to another sharing similar subsequences.
At the Edinburgh Genome Foundry, we use GeneBlocks to optimize sequence assembly, explore sets of non-annotated sequences, or visualize the differences between different versions of a sequence, and re-annotate records coming from third parties such as DNA manufacturers.
Transfer Genbank features between records
The CommonBlocks feature requires NCBI BLAST+. On Ubuntu, install it with
apt-get install ncbi-blast+You can install GeneBlocks through PIP:
pip install geneblocksAlternatively, you can unzip the sources in a folder and type:
python setup.py installfrom geneblocks import CommonBlocks
# Input sequences are in a dictionary as follows:
sequences = {'seq1': 'ATTTGCGT', 'seq2': 'ATGCCCGCACG',} # etc
common_blocks = CommonBlocks.from_sequences(sequences)
# PLOT THE BLOCKS
axes = common_blocks.plot_common_blocks()
axes[0].figure.savefig("basic_example.png", bbox_inches="tight")
# GET ALL COMMON BLOCKS AS BIOPYTHON RECORDS
blocks_records = common_blocks.common_blocks_records()
# WRITE ALL COMMON BLOCKS INTO A CSV SPREADSHEET
common_blocks.common_blocks_to_csv(target_file="blocks.csv")Result:
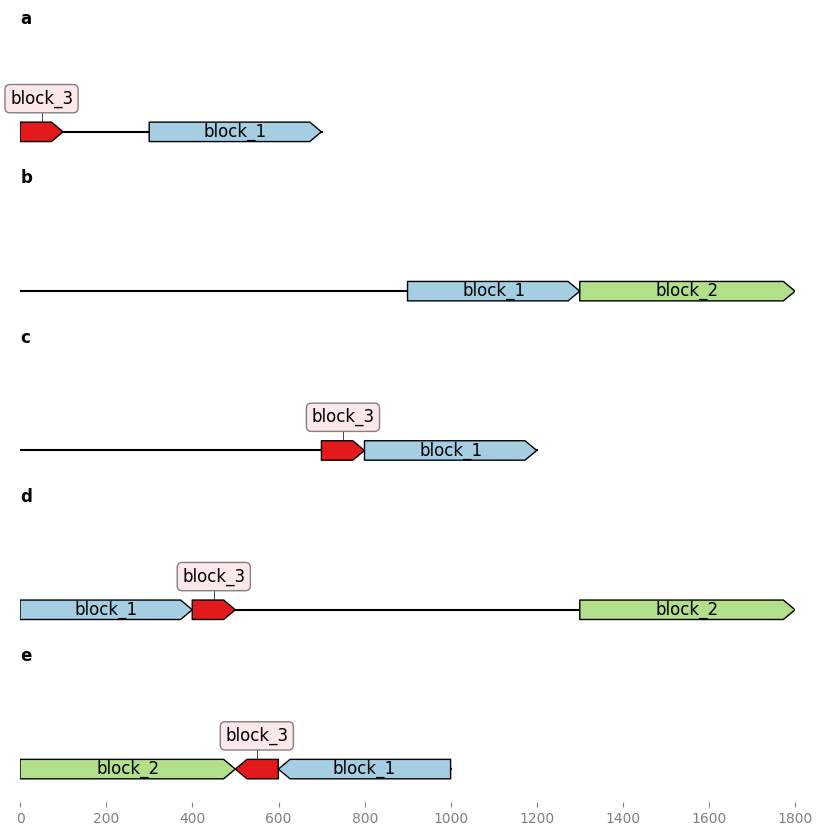
In this snippet we assume that we have two genbank records:
- A record of an annotated part, containing an expression module.
- A record of a plasmid which contains the part but the part was not properly annotated.
We will use Geneblocks to automatically detect where the part is located in the plasmid and automatically copy the features from the part record to the plasmid record.
from geneblocks import CommonBlocks, load_record
part = load_record('part.gb', name='insert')
plasmid = load_record('part.gb', name='plasmid')
blocks = CommonBlocks.from_sequences([part, plasmid])
new_records = blocks.copy_features_between_common_blocks(inplace=False)
annotated_plasmid = new_records['plasmid'] # Biopython recordThe resulting annotated plasmids has annotations from both the original plasmid and the annotated part:
![]()
seq_1 = load_record("sequence1.gb")
seq_2 = load_record("sequence2.gb")
diff_blocks = DiffBlocks.from_sequences(seq_1, seq_2)
ax1, ax2 = diff_blocks.merged().plot(figure_width=8)
ax1.figure.savefig("diff_blocks.png")Result:
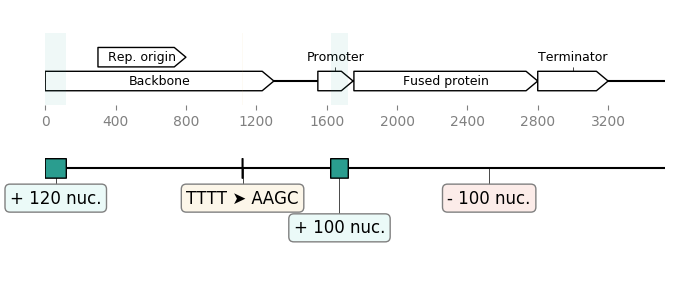
In the example below we build two ~50kb "sister" sequences with many insertions, deletions, transpositions between them, and even a self-homology. Then we ask Geneblocks to compare the two sequences, in term of common blocks, and in terms of modifications brought to the second sequence:
from geneblocks import DiffBlocks, CommonBlocks, random_dna_sequence
import geneblocks.sequence_modification_utils as smu
import matplotlib.pyplot as plt
# GENERATE 2 "SISTER" SEQUENCES FOR THE EXAMPLE
seq1 = random_dna_sequence(50000)
seq1 = smu.copy(seq1, 25000, 30000, 50000)
seq2 = seq1
seq2 = smu.insert(seq2, 39000, random_dna_sequence(100))
seq2 = smu.insert(seq2, 38000, random_dna_sequence(100))
seq2 = smu.reverse(seq2, 30000, 35000)
seq2 = smu.swap(seq2, (30000, 35000), (45000, 480000))
seq2 = smu.delete(seq2, 20000, 2000)
seq2 = smu.insert(seq2, 10000, random_dna_sequence(2000))
seq2 = smu.insert(seq2, 0, 1000*"A")
# FIND COMMON BLOCKS AND DIFFS
common_blocks = CommonBlocks.from_sequences({'seq1': seq1, 'seq2': seq2})
diff_blocks = DiffBlocks.from_sequences(seq1, seq2).merged()
# PLOT EVERYTHING
fig, axes = plt.subplots(3, 1, figsize=(15, 8))
common_blocks.plot_common_blocks(axes=axes[:-1])
diff_blocks.plot(ax=axes[-1], separate_axes=False)
axes[-1].set_xlabel("Changes in seq2 vs. seq1")
fig.savefig("complex_sequences.png", bbox_inches='tight')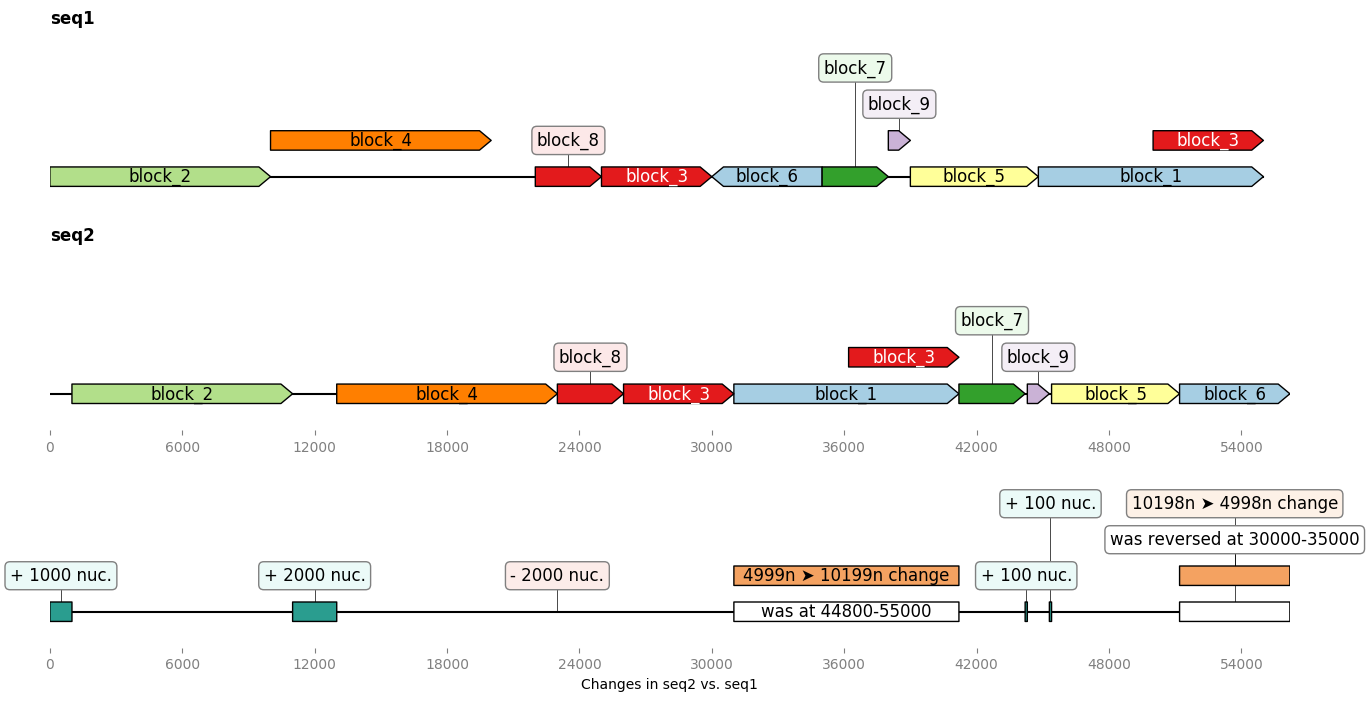
Geneblocks is an open-source software originally written at the Edinburgh Genome Foundry by Zulko and released on Github under the MIT license (Copyright 2017 Edinburgh Genome Foundry). Everyone is welcome to contribute!