

![]()


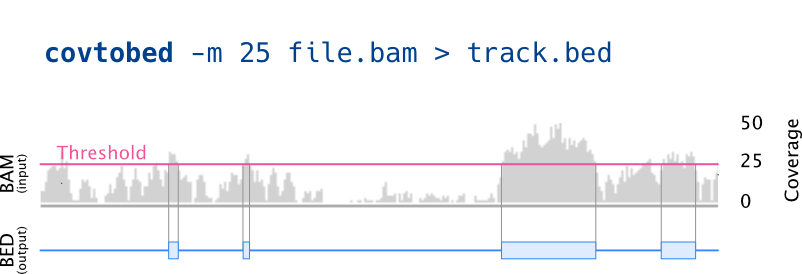
Reads one (or more) alignment files (sorted BAM) and prints a BED with the coverage. It will join consecutive bases with the same coverage, and can be used to only print a BED file with the regions having a specific coverage range.
📖 Read more in the wiki - this is the main documentation source
Features:
- Can read (sorted) BAMs from stream (like
bwa mem .. | samtools view -b | samtools sort - | covtobed) - Can print strand specific coverage to check for strand imbalance
- Can print the physical coverage (with paired-end or mate-paired libraries)
ℹ️ For more features, check the BamToCov suite.
📖 The complete documentation is available in the GitHub wiki.
Synopsis:
Usage: covtobed [options] [BAM]...
Computes coverage from alignments
Options:
-h, --help show this help message and exit
--version show program's version number and exit
--physical-coverage compute physical coverage (needs paired alignments in input)
-q MINQ, --min-mapq=MINQ
skip alignments whose mapping quality is less than MINQ
(default: 0)
-m MINCOV, --min-cov=MINCOV
print BED feature only if the coverage is bigger than
(or equal to) MINCOV (default: 0)
-x MAXCOV, --max-cov=MAXCOV
print BED feature only if the coverage is lower than
MAXCOV (default: 100000)
-l MINLEN, --min-len=MINLEN
print BED feature only if its length is bigger (or equal
to) than MINLELN (default: 1)
-z MINCTG, --min-ctg-len=MINCTG
skip reference sequences having size less or equal to MINCTG
-d, --discard-invalid-alignments
skip duplicates, failed QC, and non primary alignment,
minq>0 (or user-defined if higher) (default: 0)
--output-strands output coverage and stats separately for each strand
--format=CHOICE output format
Command:
covtobed -m 0 -x 5 test/demo.bam
Output:
[...]
NC_001416.1 0 2 0
NC_001416.1 2 6 1
NC_001416.1 6 7 2
NC_001416.1 7 12 3
NC_001416.1 12 18 4
NC_001416.1 169 170 4
NC_001416.1 201 206 4
[...]
See the full example output from different tools 📂 here
- To install with Miniconda:
conda install -c bioconda covtobed- Both covtobed, and the legacy program coverage are available as a single Docker container available from Docker Hub
:

sudo docker pull andreatelatin/covtobed
sudo docker run --rm -ti andreatelatin/covtobed coverage -h- Download Singularity image by
singularity pull docker://andreatelatin/covtobed, then:
singularity exec covtobed.simg coverage -hWhen invoked without arguments, covtobed will print a message to inform the user that it
is waiting for input from STDIN.
To suppress this message, set the environment variable COVTOBED_QUIET to 1.
covtobed is generally faster than bedtools. More details are in the benchmark page.
This tool requires libbamtools and zlib.
To manually compile:
c++ -std=c++11 *.cpp -I/path/to/bamtools/ -L${HOME}/path/to/lib/ -lbamtools -o covtobed
- This program will read the coverage from sorted BAM files. The CRAM format is not supported at the moment.
- If you find a problem feel free to raise an issue, we will try to address it as soon as possible
- Contributions are welcome via PR.
This tools uses libbamtools by Derek Barnett, Erik Garrison, Gabor Marth and Michael Stromberg, and cpp-optparse by Johannes Weißl. Both tools and this program are released with MIT license.
Giovanni Birolo (@gbirolo), University of Turin, and Andrea Telatin (@telatin), Quadram Institute Bioscience.
This program was finalized with a Flexible Talent Mobility Award funded by BBSRC through the Quadram Institute.
If you use this tool, we would really appreciate if you will cite its paper:
Releases after 1.3 (inclusive):
Giovanni Birolo, Andrea Telatin, BamToCov: an efficient toolkit for sequence coverage calculations, Bioinformatics, 2022
Releases up to 1.2:
Birolo et al., (2020). covtobed: a simple and fast tool to extract coverage tracks from BAM files. Journal of Open Source Software, 5(47), 2119, https://doi.org/10.21105/joss.02119