
CutSeq is a tool that provides an efficient wrapper for the cutadapt tool, which is powerful in handling various types of NGS libraries. Due to the complexities involved in NGS library preparation methods, mutiple operations are necessary to process sequencing reads correctly.
Take SMARTer® Stranded Total RNA-Seq Kit v3 as an example, at least 9 operations are required.

For Read 1:
- Remove the Illumina p7 adapter from the end of the sequence.
- Remove 14 nt (8+3+3) at the rightmost position of the sequence, representing UMI and linker sequence from the beginning of read 2. This is required when the library insert size is shorter than the sequencing length.
- Remove poly-T sequences at the beginning of the sequence (read 1 is oriented in reverse to the RNA, hence a polyA tail appears as a leading polyT sequence).
- Remove low-quality bases from right to left.
For Read 2:
- Remove the reverse complement Illumina p5 adapter from the end of the sequence.
- Extract the 8 nt UMI sequence from the beginning of the sequence and append it to the read name for downstream analysis.
- Mask a 6 nt linker sequence at the leftmost position immediately after clipping the UMI sequence.
- Remove poly-A sequences at the end of the read.
- Remove low-quality bases from right to left.
These operations must be performed in the correct order. The limitations of the cutadapt tool make it challenging to configure these operations in a single command, often leading to errors unnoticed in some publications.
To solve this by using cutadapt, we can run multiple cutadpat insitent sequentially or pipe multiple commands together. But this waste lots of IO and computational resource. I am thinking there a more eligent API to make things easy. Then comes this toy project. -- What you need is only one parameter which spcific what the library would looks like.
CutSeq overcomes these limitations by enabling multiple operations in a automatical manner to ensure accuracy and efficiency.
pip install cutseqExecute adapter trimming by providing a single parameter and your input files:
cutseq -A TAKARAV3 test_R1.fq.gz test_R2.fq.gzAlternatively, you can specify a custom adapter sequence:
cutseq -a "ACACGACGCTCTTCCGATCTXXX<XXXXXXNNNNNNNNAGATCGGAAGAGCACACGTC"
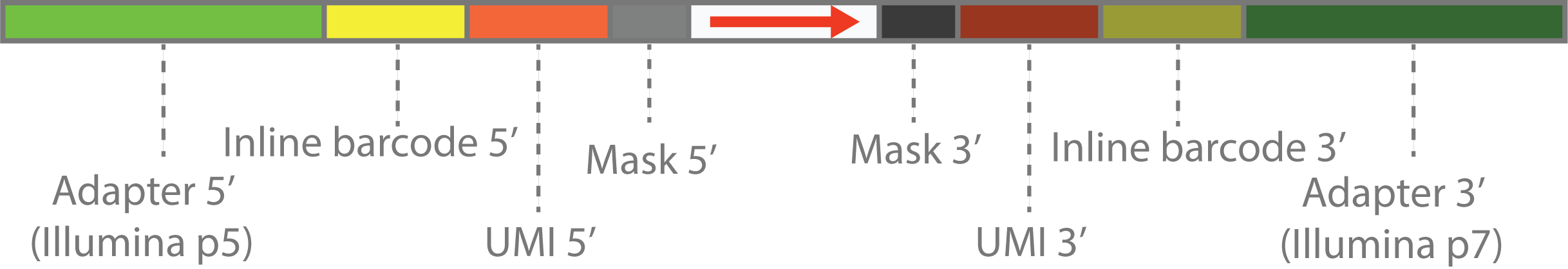
The customized scheme can be explained by diagram above.
- The outmost parts on both ends are the Illumina adapters.
- The first inner parts are inline barcode sequence or customized PCR primers in the library construction step. These are also fixed DNA sequence, and will be represented by by sequence within
(and). - The second inner parts are the UMI sequence, which is a random sequence and will be represented by
N. - The innermost parts are sequnce to be masked, which will be represented by
X. This can be random tail in the library construction step, caused by template switching or other reasons. - The center parts are the actual library sequence, which will be represented by
>,<or-.>means that sequence is forward,<means that sequence is reverse,-means that sequence orientation is unknown.
More details can be found in the document
[ ] support more library scheme