![]()
![]()


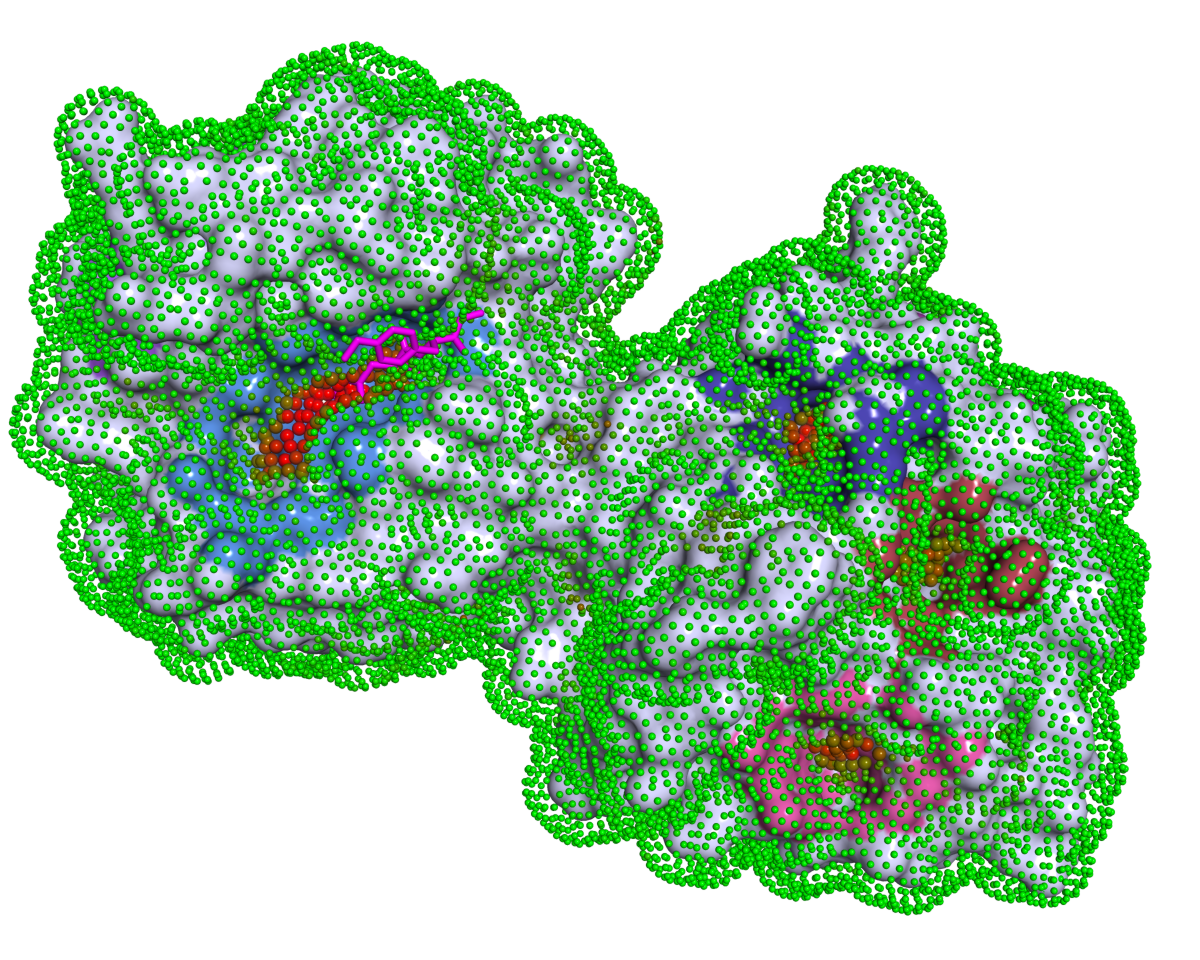
P2Rank is a stand-alone command-line program for fast and accurate prediction of ligand-binding sites from protein structures. It achieves high prediction success rates without relying on external software for computation of complex features or on a database of known protein-ligand templates.
- Version 2.5 brings speed optimizations (~2x faster prediction), ChimeraX visualizations, and improvements to rescoring (
fpocket-rescorecommand). - Version 2.4.2 adds support for BinaryCIF (
.bcif) input and rescoring of fpocket predictions in.cifformat. - Version 2.4 adds support for mmCIF (
.cif) input and contains a special profile for predictions on AlphaFold models and NMR/cryo-EM structures.
- Java 17 to 23
- PyMOL/ChimeraX for viewing visualizations (optional)
P2Rank is tested on Linux, macOS, and Windows.
P2Rank requires no installation. Binary packages are available as GitHub Releases.
- Download: https://github.com/rdk/p2rank/releases
- Source code: https://github.com/rdk/p2rank
- Datasets: https://github.com/rdk/p2rank-datasets
prank predict -f test_data/1fbl.pdb # predict pockets on a single pdb file
See more usage examples below...
P2Rank makes predictions by scoring and clustering points on the protein's solvent accessible surface. Ligandability score of individual points is determined by a machine learning model trained on a dataset of known protein-ligand complexes. For more details, see the slides and publications.
Presentation slides introducing the original version of the algorithm: Slides (pdf)
If you use P2Rank, please cite relevant papers:
- Software article about P2Rank pocket prediction tool
Krivak R, Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics. 2018 Aug. - A new web-server article about updates in the web interface prankweb.cz
Jakubec D, Skoda P, Krivak R, Novotny M, Hoksza D. PrankWeb 3: accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Research, Volume 50, Issue W1, 5 July 2022, Pages W593–W597 - Web-server article introducing the web interface at prankweb.cz
Jendele L, Krivak R, Skoda P, Novotny M, Hoksza D. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Research, Volume 47, Issue W1, 02 July 2019, Pages W345-W349 - Conference paper introducing P2Rank prediction algorithm
Krivak R, Hoksza D. P2RANK: Knowledge-Based Ligand Binding Site Prediction Using Aggregated Local Features. International Conference on Algorithms for Computational Biology 2015 Aug 4 (pp. 41-52). Springer - Research article about PRANK rescoring algorithm (now included in P2Rank)
Krivak R, Hoksza D. Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features. Journal of Cheminformatics. 2015 Dec.
Following commands can be executed in the installation directory.
prank help # print help for main commands and parameters
prank -v # print version and some system infoprank predict test.ds # run on dataset containing a list of pdb/cif files
prank predict -f test_data/1fbl.pdb # run on a single pdb file
prank predict -f test_data/1fbl.cif # run on a single mmCIF file
prank predict -f test_data/1fbl.bcif # run on a single BinaryCIF file
prank predict -f test_data/1fbl.pdb.gz # run on a single gzipped pdb file (other formats can be compressed too)
prank predict -f test_data/1fbl.cif.zst # run on a single cif file compressed with Zstandard
prank predict -threads 8 test.ds # specify num. of working threads for parallel dataset processing
prank predict -o output_here test.ds # explicitly specify output directory
prank predict -c alphafold test.ds # use alphafold config and model (config/alphafold.groovy)
# this profile is recommended for AlphaFold models, NMR and cryo-EM
# structures since it doesn't depend on b-factor as a feature For each structure file {struct_file} in the dataset, P2Rank generates several output files:
{struct_file}_predictions.csv: lists predicted pockets in order of score, including each pocket's score, center coordinates, adjacent residues, adjacent protein surface atoms, and a calibrated probability of being a ligand-binding site.{struct_file}_residues.csv: lists all residues from the input protein along with their scores, mapping to predicted pockets, and a calibrated probability of being a ligand-binding residue.- PyMol and ChimeraX visualizations:
.pmland.cxcscripts invisualizations/directory with additional files indata/.- Optional settings:
- Use
-visualizations 0to disable visualization generation. - Use
-vis_renderers 'pymol,chimerax'to toggle specific renderers on/off. - Use
-vis_copy_proteins 0to prevent copying protein structures to the visualizations directory (faster, but visualizations won't be portable).
- Use
- Optional settings:
- SAS points data: coordinates and ligandability scores for solvent-accessible surface (SAS) points are saved in
visualizations/data/{struct_file}_points.pdb.gz. Here:- Residue sequence number (position 23-26) represents the pocket rank (0 indicates no pocket).
- B-factor column contains predicted ligandability score.
You can override the default parameter values in a custom config file:
prank predict -c config/example.groovy test.ds
prank predict -c example test.ds # same effect, config/ is default location and .groovy implicit extensionIt is also possible to override parameters on the command line using their full name after - (not --).
prank predict -visualizations 0 -threads 8 test.ds # turn off visualizations and set the number of threads
prank predict -c example.groovy -visualizations 0 -threads 8 test.ds # overrides defaults as well as values from example.groovyP2Rank has many configurable parameters.
To see the list of standard parameters look into config/default.groovy and other example config files in this directory.
To see the complete commented list of all (including undocumented)
parameters see Params.groovy in the source code.
In addition to predicting new ligand binding sites, P2Rank is also able to rescore pockets predicted by other methods (Fpocket, ConCavity, SiteHound, MetaPocket2, LISE, DeepSite, and PUResNetV2.0 are supported at the moment).
Rescoring output:
{struct_file}_rescored.csv: list of pockets sorted by the new score{struct_file}_predictions.csv: same as withprank predict(since 2.5)- Note: probability column is calibrated for rescoring fpocket predictions
- visualizations
prank rescore fpocket.ds
prank rescore fpocket.ds -o output_here # explicitly specify output directory
prank rescore fpocket.ds -c rescore_2024 # use new experimental rescoring model (recommended for alphafold models)For rescoring, the dataset file needs to have a specific 2-column format. See examples in test_data/: fpocket.ds, concavity.ds, puresnet.ds.
New experimental rescoring model -c rescore_2024 shows promising result but hasn't been fully evaluated yet. It is recommended for AlphaFold models, NMR and cryo-EM structures since it doesn't depend on b-factor as a feature.
You can use fpocket-rescore command to run Fpocket and then rescore its predictions automatically.
prank fpocket-rescore test.ds # expects 'fpocket' command in PATH
prank fpocket-rescore test.ds -fpocket_command "/bin/fpocket -w m" # specify custom fpocket command (optionally with arguments)
prank fpocket-rescore test.ds -fpocket_keep_output 0 # delete fpocket output filesIn this case, the dataset file can be a simple list of pdb/cif files since Fpocket predictions will be calculated ad-hoc.
prank fpocket-rescore will produce predictions.csv as well, so it can be used as an in-place replacement for prank predict in most scenarios.
Note: if you use fpocket-rescore, please cite Fpocket as well.
Use following commands to calculate prediction metrics (prediction success rates using DCA, DCC, ...) on structure files, where the ligands are present.
prank eval-predict -f test_data/1fbl.pdb # evaluate default prediction model on a single file
prank eval-predict test.ds # evaluate default prediction model on a dataset with known ligands
prank eval-predict -c alphafold test.ds # evaluate specific prediction model on a dataset with known ligands
prank eval-rescore fpocket.ds # evaluate default rescoring model on a dataset with known ligands
prank eval-rescore -c rescore_2024 fpocket.ds # evaluate specific rescoring model on a dataset with known ligandsThis project uses Gradle build system via included Gradle wrapper.
On Windows, use bash to run build commands (installed by default with Git for Windows).
git clone https://github.com/rdk/p2rank.git && cd p2rank
./make.sh
./unit-tests.sh # optionally you can run tests to check everything works fine on your machine
./tests.sh quick # runs further testsNow you can run the program via:
distro/prank # standard mode that is run in production
./prank.sh # development/training mode To use ./prank.sh (development/training mode) first you need to copy and edit misc/locval-env.sh into repo root directory (see training tutorial).
Fpocket is a widely used open source ligand binding site prediction program. It is fast, easy to use and well documented. As such, it served as a great inspiration for this project.
Some practical differences:
- Fpocket
- has a much smaller memory footprint
- runs faster when executed on a single protein
- produces a high number of less relevant pockets (and since the default scoring function isn't very effective, the most relevant pockets often don't get to the top)
- contains MDpocket algorithm for pocket predictions from molecular trajectories
- still better documented
- P2Rank
- achieves significantly higher identification success rates when considering top-ranked pockets
- produces a smaller number of more relevant pockets
- speed:
- slower when running on a single protein (due to JVM startup cost)
- approximately as fast on average running on a big dataset on a single core
- due to parallel implementation potentially much faster on multi-core machines
- higher memory footprint (~1G but doesn't grow much with more parallel threads)
Both Fpocket and P2Rank have many configurable parameters that influence behaviour of the algorithm and can be tweaked to achieve better results for particular requirements.
This program builds upon software written by other people, either through library dependencies or through code included in its source tree (where no library builds were available). Notably:
- FastRandomForest by Fran Supek (https://code.google.com/archive/p/fast-random-forest/)
- FastRandomForest 2.0 (https://github.com/GenomeDataScience/FastRandomForest)
- KDTree by Rednaxela (http://robowiki.net/wiki/User:Rednaxela/kD-Tree)
- BioJava (https://github.com/biojava)
- Chemistry Development Kit (https://github.com/cdk)
- Weka (http://www.cs.waikato.ac.nz/ml/weka/)
We welcome any bug reports, enhancement requests, and other contributions. To submit a bug report or enhancement request, please use the GitHub issues tracker. For more substantial contributions, please fork this repo, push your changes to your fork, and submit a pull request with a good commit message.