
![]()



lineage provides a framework for analyzing genotype (raw data) files from direct-to-consumer
(DTC) DNA testing companies, primarily for the purposes of genetic genealogy.
- Find shared DNA and genes between individuals
- Compute centiMorgans (cMs) of shared DNA using a variety of genetic maps (e.g., HapMap Phase II, 1000 Genomes Project)
- Plot shared DNA between individuals
- Find discordant SNPs between child and parent(s)
- Read, write, merge, and remap SNPs for an individual via the snps package
lineage supports all genotype files supported by snps.
lineage is available on the
Python Package Index. Install lineage (and its required
Python dependencies) via pip:
$ pip install lineage
Also see the installation documentation.
lineage requires Python 3.8+ and the following Python packages:
Import Lineage and instantiate a Lineage object:
>>> from lineage import Lineage
>>> l = Lineage()First, let's setup logging to get some helpful output:
>>> import logging, sys
>>> logger = logging.getLogger()
>>> logger.setLevel(logging.INFO)
>>> logger.addHandler(logging.StreamHandler(sys.stdout))Now we're ready to download some example data from openSNP:
>>> paths = l.download_example_datasets()
Downloading resources/662.23andme.340.txt.gz
Downloading resources/662.ftdna-illumina.341.csv.gz
Downloading resources/663.23andme.305.txt.gz
Downloading resources/4583.ftdna-illumina.3482.csv.gz
Downloading resources/4584.ftdna-illumina.3483.csv.gzWe'll call these datasets User662, User663, User4583, and User4584.
Create an Individual in the context of the lineage framework to interact with the
User662 dataset:
>>> user662 = l.create_individual('User662', ['resources/662.23andme.340.txt.gz', 'resources/662.ftdna-illumina.341.csv.gz'])
Loading SNPs('662.23andme.340.txt.gz')
Merging SNPs('662.ftdna-illumina.341.csv.gz')
SNPs('662.ftdna-illumina.341.csv.gz') has Build 36; remapping to Build 37
Downloading resources/NCBI36_GRCh37.tar.gz
27 SNP positions were discrepant; keeping original positions
151 SNP genotypes were discrepant; marking those as nullHere we created user662 with the name User662. In the process, we merged two raw data
files for this individual. Specifically:
662.23andme.340.txt.gzwas loaded.- Then,
662.ftdna-illumina.341.csv.gzwas merged. In the process, it was found to have Build 36. So, it was automatically remapped to Build 37 (downloading the remapping data in the process) to match the build of the SNPs already loaded. After this merge, 27 SNP positions and 151 SNP genotypes were found to be discrepant.
user662 is represented by an Individual object, which inherits from snps.SNPs.
Therefore, all of the properties and methods
available to a SNPs object are available here; for example:
>>> len(user662.discrepant_merge_genotypes)
151
>>> user662.build
37
>>> user662.build_detected
True
>>> user662.assembly
'GRCh37'
>>> user662.count
1006960As such, SNPs can be saved, remapped, merged, etc. See the snps package for further examples.
Let's create another Individual for the User663 dataset:
>>> user663 = l.create_individual('User663', 'resources/663.23andme.305.txt.gz')
Loading SNPs('663.23andme.305.txt.gz')Now we can perform some analysis between the User662 and User663 datasets.
First, let's find discordant SNPs (i.e., SNP data that is not consistent with Mendelian inheritance):
>>> discordant_snps = l.find_discordant_snps(user662, user663, save_output=True)
Saving output/discordant_snps_User662_User663_GRCh37.csvAll output files are saved to
the output directory (a parameter to Lineage).
This method also returns a pandas.DataFrame, and it can be inspected interactively at
the prompt, although the same output is available in the CSV file.
>>> len(discordant_snps.loc[discordant_snps['chrom'] != 'MT'])
37Not counting mtDNA SNPs, there are 37 discordant SNPs between these two datasets.
lineage uses the probabilistic recombination rates throughout the human genome from the
International HapMap Project
and the 1000 Genomes Project to compute the shared DNA
(in centiMorgans) between two individuals. Additionally, lineage denotes when the shared DNA
is shared on either one or both chromosomes in a pair. For example, when siblings share a segment
of DNA on both chromosomes, they inherited the same DNA from their mother and father for that
segment.
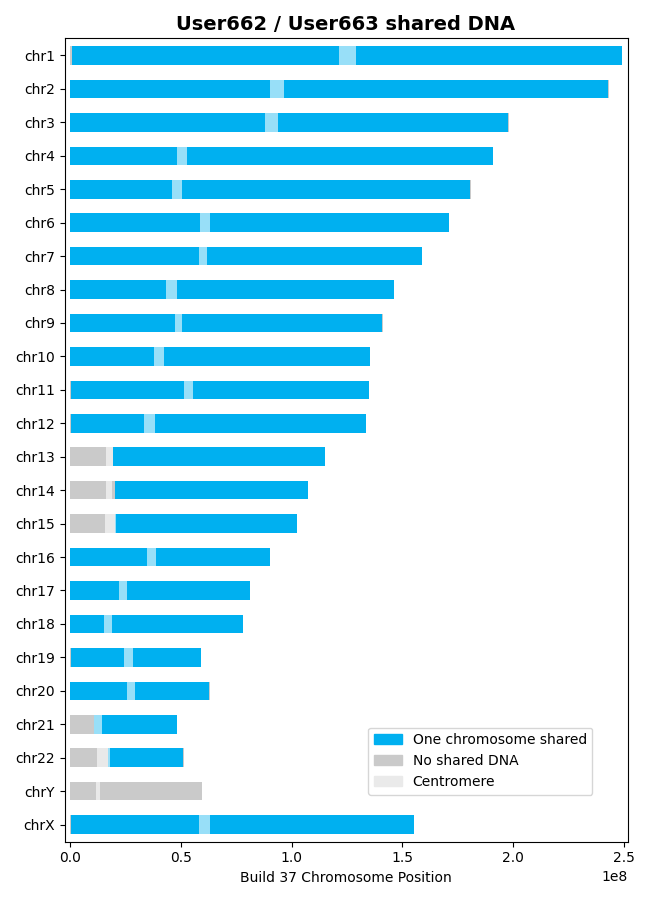
With that background, let's find the shared DNA between the User662 and User663 datasets,
calculating the centiMorgans of shared DNA and plotting the results:
>>> results = l.find_shared_dna([user662, user663], cM_threshold=0.75, snp_threshold=1100)
Downloading resources/genetic_map_HapMapII_GRCh37.tar.gz
Downloading resources/cytoBand_hg19.txt.gz
Saving output/shared_dna_User662_User663_0p75cM_1100snps_GRCh37_HapMap2.png
Saving output/shared_dna_one_chrom_User662_User663_0p75cM_1100snps_GRCh37_HapMap2.csvNotice that the centiMorgan and SNP thresholds for each DNA segment can be tuned. Additionally,
notice that two files were downloaded to facilitate the analysis and plotting - future analyses
will use the downloaded files instead of downloading the files again. Finally, notice that a list
of individuals is passed to find_shared_dna... This list can contain an arbitrary number of
individuals, and lineage will find shared DNA across all individuals in the list (i.e.,
where all individuals share segments of DNA on either one or both chromosomes).
Output is returned as a dictionary with the following keys (pandas.DataFrame and
pandas.Index items):
>>> sorted(results.keys())
['one_chrom_discrepant_snps', 'one_chrom_shared_dna', 'one_chrom_shared_genes', 'two_chrom_discrepant_snps', 'two_chrom_shared_dna', 'two_chrom_shared_genes']In this example, there are 27 segments of shared DNA:
>>> len(results['one_chrom_shared_dna'])
27Also, output files are
created; these files are detailed in the documentation and their generation can be disabled with a
save_output=False argument. In this example, the output files consist of a CSV file that
details the shared segments of DNA on one chromosome and a plot that illustrates the shared DNA:
The Central Dogma of Molecular Biology states that genetic information flows from DNA to mRNA to proteins: DNA is transcribed into mRNA, and mRNA is translated into a protein. It's more complicated than this (it's biology after all), but generally, one mRNA produces one protein, and the mRNA / protein is considered a gene.
Therefore, it would be interesting to understand not just what DNA is shared between individuals,
but what genes are shared between individuals with the same variations. In other words,
what genes are producing the same proteins? [*] Since lineage can determine the shared DNA
between individuals, it can use that information to determine what genes are also shared on
either one or both chromosomes.
| [*] | In theory, shared segments of DNA should be producing the same proteins, but there are many complexities, such as copy number variation (CNV), gene expression, etc. |
For this example, let's create two more Individuals for the User4583 and User4584
datasets:
>>> user4583 = l.create_individual('User4583', 'resources/4583.ftdna-illumina.3482.csv.gz')
Loading SNPs('4583.ftdna-illumina.3482.csv.gz')>>> user4584 = l.create_individual('User4584', 'resources/4584.ftdna-illumina.3483.csv.gz')
Loading SNPs('4584.ftdna-illumina.3483.csv.gz')Now let's find the shared genes, specifying a population-specific 1000 Genomes Project genetic map (e.g., as predicted by ezancestry!):
>>> results = l.find_shared_dna([user4583, user4584], shared_genes=True, genetic_map="CEU")
Downloading resources/CEU_omni_recombination_20130507.tar
Downloading resources/knownGene_hg19.txt.gz
Downloading resources/kgXref_hg19.txt.gz
Saving output/shared_dna_User4583_User4584_0p75cM_1100snps_GRCh37_CEU.png
Saving output/shared_dna_one_chrom_User4583_User4584_0p75cM_1100snps_GRCh37_CEU.csv
Saving output/shared_dna_two_chroms_User4583_User4584_0p75cM_1100snps_GRCh37_CEU.csv
Saving output/shared_genes_one_chrom_User4583_User4584_0p75cM_1100snps_GRCh37_CEU.csv
Saving output/shared_genes_two_chroms_User4583_User4584_0p75cM_1100snps_GRCh37_CEU.csvThe plot that illustrates the shared DNA is shown below. Note that in addition to outputting the shared DNA segments on either one or both chromosomes, the shared genes on either one or both chromosomes are also output.
Note
Shared DNA is not computed on the X chromosome with the 1000 Genomes Project genetic maps since the X chromosome is not included in these genetic maps.
In this example, there are 15,976 shared genes on both chromosomes transcribed from 36 segments of shared DNA:
>>> len(results['two_chrom_shared_genes'])
15976
>>> len(results['two_chrom_shared_dna'])
36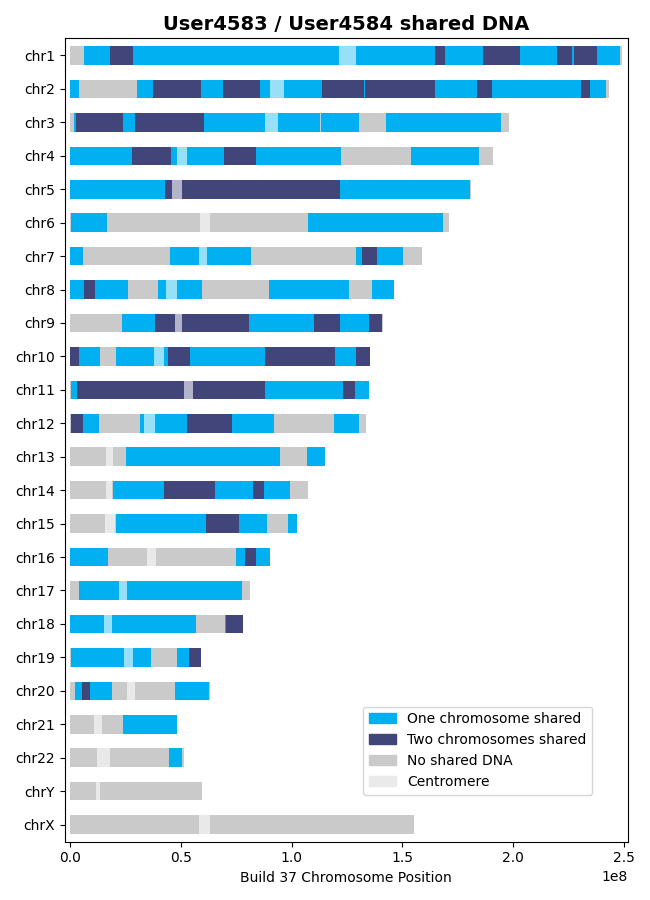
Documentation is available here.
Thanks to Whit Athey, Ryan Dale, Binh Bui, Jeff Gill, Gopal Vashishtha, CS50, and openSNP.
lineage incorporates code and concepts generated with the assistance of
OpenAI's ChatGPT . ✨